The Effect of the Time of Zygote Vitrification on the Success Rate of Frozen-Thawed IVF/ICSI Cycles
The Effect of the Time of Zygote Vitrification on the Success Rate of Frozen-Thawed IVF/ICSI Cycles
Saurabh *1, Hardeep Singh 2
1. Chief Embryologist, Jammu-180001, J & K, India.
2. Embryologist, Jammu-180001, J & K, India.
*Correspondence to: Saurabh, Chief Embryologist, Jammu-180001, J & K, India.
Copyright
© 2023 Saurabh. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 30 August 2023
Published: 15 September 2023
Abstract
Introduction: Cryopreservation of embryonic zygotes at the subsequent pronucleus (2PN) stage is an important part of the in vitro fertilization science in countries with strict rules about how to grow eggs. The vitrification of created eggs is a good way to handle work when resources are limited, and it works with a wide range of infertile treatment plans, no matter how they are made. Vitrification of fertilized eggs has resulted in good survival and good clinical outcomes, but the optimal time to start is still unknown, but is often done between 17 and 21 hours after insemination (hpi). To contribute to the standardization of cryopreservation of fertilized oocytes, we sought to elucidate the relationship between the pregnancy rate and the time between insemination (in vitro fertilization and microinsemination) and vitrification.
Methods. We retrospectively reviewed data on pregnancy outcomes and timing of vitrification collected between 2011 and 2019. After heating the vitrified zygotes to stage 2PN, all participating women underwent embryo transfer.
Results. 182 (38.9%) of the 468 embryo transfers led to pregnancy, whereas 286 (61.1%) did not. The pregnant group's mean HPI was 18.74 0.63, whereas the non-pregnant group's HPI was 18.62 0.64 (OR 1.36, 95% CI 1.01; 1, 83, p=0.045). Adjusted for uterine illness, mother age, AMH, IBD oocyte weight, history of successful pregnancies, endometriosis, ROS, nicotine use, and male infertility, the time of vitrification has an impact on the rate of conception. No predictive impact was shown. Between insemination and vitrification, three time windows of 5:00–6:00 p.m. (group A), 6:01–7:00 p.m. (group B), and 7:01–9:00 p.m. (group C) were established.
Discussion: To improve the quality of long-term ART results that show sexuality, the cryopreservation of zygotes from IVF/ICSI rounds needs to be standardized. Because of these results, it would be good to learn more about how the time of vitrification affects clinical outcomes.
keywords: Cryopreservation, zygote vitrification, IVF/ICSI, pregnancy rate, schedule, retrospective analysis, embryo transfer, ART results.
The Effect of the Time of Zygote Vitrification on the Success Rate of Frozen-Thawed IVF/ICSI Cycles
1.Introduction
Even though there is a continuing trend toward longer embryo culture (Al-Hasani et al., 2007), it is still important for IVF labs to freeze bipronuclear (2PN) stage zygotes. Nikolatos and Al-Hasani (2000) and Vanderzwalmen et al. (2006) say that 2PN stage cryopreservation is a strong support for a wide range of treatment conditions related to extra embryo saving and freeze-all indications. The use of 2PN cryopreservation has been debated for a number of reasons, such as the mother's old age, a low number of zygotes, the need for cleavage stage transfer, and a history of no blastulation. Some IVF labs freeze embryos during the 2PN stage to help with logistics and work load management. This makes the problem worse. Montag et al. (2006) say that zygote cryopreservation can be used to get around legal limits on how many zygotes can be kept in culture and on how long embryos can be kept in culture. Germany only lets people grow three zygotes if the most eggs that can be moved are three. So, it is required by law to keep zygotes that aren't meant to be transferred. Swiss law has similar restrictions. It limits the number of zygotes that can develop to 12. In many centers, it is important to cryopreserve bipronuclear levels.
Golakov et al. (2018) found that bipronuclear stage cryopreservation has above 90% survival rates after warming. This is because there is no spindle machinery, the stage is unicellular, and it is easy to see that the stage has survived by going through syngamy and developing to the first cleavage. The one and only RCT (Shapiro et al., 2015) that looked at the beneficial effects of blastocyst cryopreservation vs. 2PN found that both were good. It's important to remember that the trial used an old method called "slow freezing." Similar methods were used in two more studies (Troup et al., 1991; Senn et al., 2000) that directly compared the results of bipronuclear stage freezing and cleavage stage freezing-thawing cycles and found that bipronuclear stage freezing worked better. Right now, vitrification is the best cryopreservation method for all stages of growth (Rienzi et al., 2017). When it comes to zygotes, vitrification depends on time because so much happens between ovulation and the first cleavage, but not when it comes to embryos that are further along.
After the sperm gets inside the egg, the egg goes through meiosis, releases the second polar body, makes male and female 2PNs, and then moves into the G1 phase of the cell cycle. (Coticchio et al., 2018) When ICSI is used, time-lapse tests have shown that 2PNs can show up as early as 6 hpi. (Capmany et al., 1996), the 2PNs continue to copy DNA in the S phase until the zygote enters the G2 phase at about 18 hpi. Balakier et al. (1993) say that zygotes must be vitrified during the G2 phase and before the 2PN breaks down (syngamy) to protect the embryo from damage caused by freezing it in a weak state. The zygote starts to get ready for mitosis during the G2 phase. This happens at 23 hpi, where 2PN breaks down (Coticchio et al., 2018).
Even though 18 to 23 hours might seem like enough time for G2, the numbers above are just guesses because of how insemination works and because some eggs grow faster or slower than others. It's not impossible for two PNs to start a syngamic link after 18 hpi. Nagy et al. (1994), van Wissen et al. (1995), Capmany et al. (1996), and Neuber et al. (2003) all found that PN fell apart between 18 and 31 hpi. Fancsovits et al. (2005) were surprised to find that PN breakdown between 22 and 25 hpi was linked to lower clinical pregnancy rates than PN breakdown before 22 hpi. Azzarello et al. (2012) found that PN faded on average 24 hpi in zygotes that gave birth to live offspring, but that PN breaking down before 21 hpi was linked to no live births.
Because of these differences in the important stages of the zygote cell cycle, it is hard to decide whether or not to cryopreserve. Wright and his partners did a study to find out what the best time was to freeze the zygotes in this case. Wright et al. (1990) found that slow-frozen zygotes that were used in IVF after 23 hpi had a 39% chance of becoming pregnant, compared to 13% before that time. Later study showed that when the stepwise freezing method was used to cryopreserve 2PN stage zygotes between 16 and 23 hpi, there was a 24% chance of getting pregnant (Miller and Goldberg, 1995). Macas et al. et al. (Macas et al., 1998; Damario et al., 1999) say, however, that zygotes can be frozen at around 20 hpi without IVF or ICSI. Other experts said that normal zygotes from IVF could be vitrified at about 17 hours post-implantation (hpi).
Given how long the G2 phase lasts and how many different times cryopreservation can be done, vitrifying 2PN zygotes between 17 and 21 hours post-fertilization seems like a good plan. At this time, however, there is no study on the link between certain times of vitrification and clinical results. As far as we know, no one has looked into the best time between 2PN vitrification and fertilization to improve the chance of getting pregnant. In order to find the best time to vitrify zygotes at the 2PN stage, we looked at how the clinical pregnancy rate changed when different amounts of time passed between fertilization and vitrification.
2. Materials and methods
2.1 Study design
This study looks at the link between the time between egg fertilization and 2PN zygote vitrification during the freeze-thaw transfer cycle and the chance of getting pregnant.All research participants experienced the transfer of one or two embryos to the University Hospital of Zurich between January 2011 and December 2019 after freezing 2PNs acquired either in vitro fertilization or micro-insemination in a rice field. A short protocol, a long protocol, or a modified version of the antagonist protocol were used to treat women. The healthcare provider invited and notified eligible women to participate in the study. Each participant gave their written permission and confirmed their participation by giving their verbal consent to the analysis of the relevant data.
2.2 Inclusion and exclusion criteria
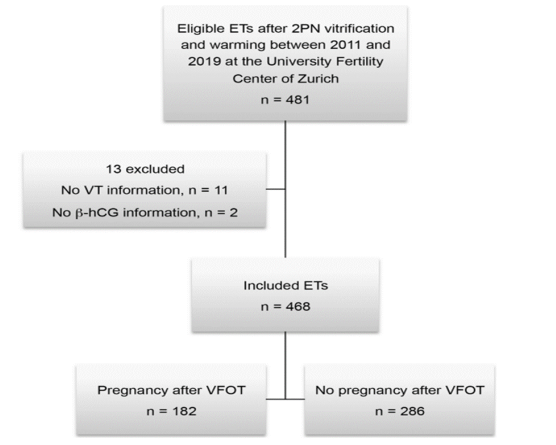
Women between the ages of 19 and 44 who had IVF provided data (mean 35.1 4.28 SD). In this review, women who knew the length of vitrification and the results of pregnancy or loss after vitrifying fertilized eggs at stage 2PN were included. Only each woman's initial embryo transfer following cryopreservation throughout the research period was utilized for the evaluation in order to gather independent data and to rule out the "learning effect" of earlier treatment as a potential confounder. There was no cleavage or blastocyst stage. Only vitrified stage 2PN zygotes were used. In this study, over 468 freeze-thaw cycles,
In fig. 1 shows the entire study cohort. Transfer after 2PN vitrification. ET, embryo transfer; VT, vitrification time
2.3 Endometrial preparation for transplantation and controlled ovarian stimulation (COS)
The short-term or blocker program gave the women a progestogen (10 mg/day) for 12–4 weeks, while the long-term COS program gave the women triptorelin (0.1 mg/day) on cycle day 21. People used recombinant FSH or HMG. Combine with short- or long-term GnRH agonist regimens or GnRH antagonist protocols for COS. Oocytes with hCG either 6500IE (Ovitrelle, Merck, Switzerland) or around 1600IE hCG under the GnRHagonist protocol after GnRHagonist supplementation if at least 3 follicles less than 17 mm in diameter were found in the vagina Stimulated terminal cell maturation. Ultrasound. Ultrasound-guided egg retrieval was performed 35 to 37 hours after the initiation dose of the hCG/GnRH antagonist. Prior to embryo transfer, progesterone 1000 mg/day was administered intravaginally after 6 mg/day oral estrogen to create an artificial endometrium. In the natural cycle, transplantation took place on the 1st or 2nd day after thawing, or on the 2nd (43-45 h) or 3rd day (67-69 h) after insemination.
2.4 Control technology
The transvaginal follicle retrieval needle (Vitrolife) was moved between the two ovaries during ultrasound-guided egg extraction. The fluid from the follicles was put in 14 mL tubes with a round bottom Aand kept warm by a heater block set to 37°C. For the search for oocytes, 60 mm Petri plates with a flat bottom and laminar flow were used. All cumulus-oocyte complexes (COCs) were grown in a humid room at 37°C and 6% CO2 with oil-soaked fertilizing medium (OVOIL, Vitrolife) (Global for The Process of fertilizing G-IVF). IVF or ICSI was used to fertilize the egg, depending on how good the sperm was. This was done 4 to 6 hours after the egg was taken out. After fertilization, eggs were grown in a MINC Benchtop incubator (COOK) using Global Total or G1 Vitrolife medium on a microdroplet plate. For ICSI, the oocytes were taken out of the egg using an enzyme called enzyme (80 IU/mL). 700 ul of G-IVF, which stands for Global for Fertilization medium in oil, was mixed with 10 106 spermatozoa/ml for IVF. In the wells of 4-well plates, this was done. After the eggs were fertilized, they were looked at 16–19 hours later to see if they had two 2PN and four polar bodies. The 2PN zygotes that were chosen for transfers on days 2 and 3 were kept in culture right away. Before vitrification, the presence of different 2PNs was checked to avoid vitrifying zygotes that were in syngamy. More 2PN zygotes were vitrified to get ready for more melting cycles.
Table 1 presents the socio-epidemiological characteristics of the groups.
Embryo transfer or ET. An independent t-test for age and BMI (ordinal variables) and a chi-square test for smoking and nationality (categorical variables) constitute the Wilcoxon-Mann-Whitney test. BMI > 24.9 kg/m2 - overweight.
2.5 Glazing
All 2PN zygotes underwent cryotopic vitrification throughout the cryopreservation process. An open vitrification supports with polyethylene strips and a protective covering is used in this procedure. Only a tiny coating of the solution that was put on the film strip after application is left on the preserved in freezing cells. The cooling and heating rates are increased by using this tiny volume to 2300°C and 4210°C, respectively (Kuwayama 2007).
Cryotope technology is almost untouched by cold because of how well vitrification works. The Cryotop® This study used the Device Open System for vitrification, Kitasato Vitrification Solution (VT601), and Kitasato The thawing Solutions (VT602). For embryo transfer, only two the zygotes during the 2PN state were placed into each cryofurnace. At 17 and 21 HPI, 2PN zygotes were frozen in liquid nitrogen.
2.6 Definitions
Metaphase II (MII) oocytes were assumed to be fully developed and viable. MII oocytes are the total amount of fertilized or infertile oocytes that happened after IVF while a definite polar body was present. In our study, the following conditions qualified for reproductive therapy: uterine and tubular diseases, primary or secondary sterility, and endo (with various rASRM stages). If a transfer in the past resulted in pregnancy, it was considered successful. Between 17:00 and 18:00 hours, from 18:01 and 19:00 hours, and between 19:01 and 21:00 hours after irradiation, three groups of vitrified zygote transfer were made. This division was created based on the amount of time between insemination and vitrification. The time it took, on a typical basis, between the beginning of the insemination operation to its finish was used to compute the vitrification time. The main discovery (biochemical and clinical) was the pregnancy rate. 14 days following embryo transfer, levels of hCG more than 10 mIU/ml were used to detect biochemical pregnancies (a negative test result was 0 mIU/mL). A transvaginal ultrasound used to diagnose clinical pregnancy found an intrauterine gestational sac to be present. According to guidelines published by the World Health Organization, the body mass index (BMI = kg/m2) was categorized. Being overweight in this research referred to a BMI of 24.9 or greater.
Table 2: Overview of indications for fertility treatment
Embryo transfer or ET. Use a chi-square test for all variables (categorical variables). Oligomenorrhea/amenorrhea with anovulation.
2.7 samples were taken
Based on a similar study (Al-Hasani et al., 2007), we thought that from 17:00 to 18:00 hpi, the number of pregnancies would drop by 25% to 30%. Cohen's h (Cohen J., 1998) was used to find out how much different the two numbers were. Using a normal power and sample size estimate, the number of subjects needed to find different effect sizes between the groups with the earliest and latest time between insemination and vitrification was found. This was done using a two-sided hypothesis test with enough statistical power (at least 80%) and a significance level of alpha =.05. At the same level of significance and 80% statistical power, a difference of 10% in the pregnancy rate between two groups that each had 329 embryo transfers would be clear. With a cohort size of n = 152, a difference of 15% in birth rates would be significant, but a difference of 20% would require 89 embryo transfers in each group. The study looked at 468 2PN zygote exchanges that happened in the womb. There were 130 people in the group with the smallest time between conception and vitrification and 57 people in the group with the greatest time between conception and vitrification. This means that the retrospective sample doesn't allow comparisons with enough statistical strength to find changes in group shares of less than 20%. Follow the steps above to figure out what the current study is about.
Table 3. Overview of prerequisites for infertility treatment.
Note:Embryo transfer or ET. Chi-square test for categorical variables and Wilcoxon-Mann-Whitney test for ordinal variables.
3. Data evaluation
The statistical tests were done with IBM SPSS version 26, which is an advanced software tool for statistical research. Absolute numbers (n), percentages (%), 95% confidence intervals, the mean, and the standard deviation are used to show the results. Statistics that describe give a numerical overview of what was seen. To compare ordinal variables between the two groups and mean values, Wilcoxon-Mann-Whitney nonparametric testing and the chi-square test were employed, respectively. When there were less than 5 individuals in the Fisher test group, exact tests were applied when comparing 2 2 crosstabulations. A two-sided test was used if required. In order to investigate the associations between the dependent variables and the outcome variables (pregnancy rate), both univariate and multivariate logistic regression analyses were conducted. Odds ratios are used to show association after adjusting for confounders. All factors were initially used in univariate analysis. The logistic multivariate score structure includes all significant variables with an alpha score of 0.05 for univariate analysis. At a significance level of α = 0.05, all hypothesis tests are considered significant.
3. Results
3.1 Verify demographic data
After 2PN zygotes were vitrified and thawed, we examined the data from 468 embryo transfers. 286 embryo transfers did not result in pregnancy, whereas a total of 182 did (Fig. 1).
The total pregnancy rate was 35.6% as a result. 139 live births came from 182 transplants with positive pregnancy tests. Careful comparisons were made between the sociodemographic characteristics of women who had successful and failed pregnancies. Table 1 demonstrates a difference in maternal age between the two groups that is statistically significant for smoking (p = 0.049) and fertility therapy (p = 0.004). Male infertility symptoms were extremely prevalent, despite the rarity of uterine illnesses and endometriosis in expectant women (Table 2). Additionally, the antral follicle count (AFC) of the pregnant women in our group was greater.
Table 4: Results of infertility treatment
Note:The table shows fertility results, including number of mature oocytes (MII), number of previous successful fresh transplants, and number of previous miscarriages. The p-value indicates the statistical significance of the observed differences between the groups.
3.2 We recommend vitrification at 19-21 hp/in.
The women who did not have successful pregnancies had a mean insemination-to-vitrification interval that was considerably longer in the first group (18.74.63 versus 18.62.64 hpi, p = 0.038; Table 5). We performed a univariate analysis utilizing the pregnancy rate and vitrification duration as constant parameters in order to extend this first result. This relationship had a p-value of 0.045, an OR of 1.36, and a 95% confidence interval of 1.01. However, it lost its statistical significance after a multivariate analysis (Table 6) that took into account changes in the groups' sociodemographic features.
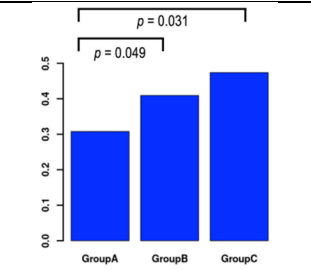
Next, we did a subset analysis based on the three time periods from conception to vitrification (Table 5) to see if we could use the timing as a condition before starting vitrification. While the pregnant group's vitrification took an average of longer (17, 94, 17 hours), it started in group A between 5:00 and 6:00 p.m. Vitrification for the pregnant women in group B started between 6 and 7 o'clock and lasted an average of 18.77 0.25 h/in. Between 7:00 and 9:30 p.m., vitrification began in group C, with the pregnant group's average onset time being 19.7–7.25 h/in. The mean intervals were the same for both pregnant and non-pregnant women in each of the three groups. Table 7 shows that group A had a pregnancy rate of 30.80%, group B had a pregnancy rate of 40.90%, and group C had a pregnancy rate of 47.40%. In univariate analysis, fertility potential was substantially linked with the three groups (OR 1.45, p = 0.018), but this association was abolished in multivariate analysis (Table 8). For univariate analysis, an OR of 2.03 (p = 0.031) was used to compare the most recent and oldest time intervals, and an OR of 1.30 (p=0.370) was used to compare the longest and intermediate groups. Time intervals (p=0.049) utilizing an OR of 1.56 and an OR of 1.30 (Table 9, Figure 2). These observations are not supported by multivariate analysis. The earliest and intermediate time periods are contrasted using an OR of 1.56 (p=0, 049) (Table 9, Figure 2). These observations are not confirmed by multivariate analysis. The difference between the middle and earliest time periods was 1.56 (p=.049) (Table 9, Fig. 2). These observations are not confirmed by multivariate analysis.
Figure 2. Comparison of success rates between groups using univariate analysis and distribution of pregnancy outcomes by time group. The vitrification time for Group A was 5:00 p.m. to 6:00 p.m. per inch, Group B - 6:01 p.m. to 7:00 p.m. per inch, and Group C - 7:01 p.m. to 9:00 p.m. per inch.
Table 5: Summary of hourly averages
NOTE. This table gives an overview of the time-averaged vitrification times in hours. The p-value indicates the statistical significance of the observed differences between the groups. Group A represents the vitrification time range from 5:00 p.m. to 6:00 p.m., group B represents the range from 6:01 p.m. to 7:00 p.m., and group C represents the range from 7:01 p.m. to 9:00 p.m.
Table 6: The vitrification time is analyzed as a continuous variable in univariate and multivariate analysis.
A. Vitrification time (continuous variable), age, uterine disease, IBD oocytes, endometriosis, and male causation were all taken into account. b A constant variable. a pregnant woman.
Note. The outcomes of univariate and multivariate analyses with vitrification time as a continuous variable are displayed in this table. There are provided approximate odds ratios (ORs) and p-values in addition to adjusted ORs and p-values that have been taken into account. p 0.05 in the raw is regarded as significant. P-values that have been corrected account for variables that could have affected the findings. Significant results are those with an adjusted p 0.05. Age, uterine pathology, AMH, IBD oocytes, successful past fresh transfer, endometriosis, AFC (antral follicle count), nicotine use, and male ancestry were among the predictors.
Table 7: Distribution of pregnancy outcomes at different insemination-vitrification time intervals.
NOTE. The distribution of pregnancies according to various lag times between insemination and vitrification is shown in the following table. Following embryo transfer (ET) and 2PN vitrification, the overall number of pregnancies and the absence of pregnancies are indicated.
Table 8: Three groups of vitrification times in univariate and multivariate analysis
A. Vitrification time (group), uterine pathology, age, AMH, IBD, endometriosis, and male factors have all been taken into account. made consists of three groups. a pregnant woman.
NOTE: The findings of the univariate and multivariate analyses using the three groups of vitrification periods are shown in this table. For every predictor, the raw odds ratio (OR), 95% confidence interval (CI), raw p-value, adjusted OR, and adjusted p-value are shown.
Table 9: Comparison of success rates by time slot
Regression's p-value (group comparison). b Vitrification time (group), uterine pathology, age, AMH, IBD, endometriosis, and male factors were all taken into account. c Fisher's exact test with two sides. d Two-degrees-of-freedom Chi-square test. NOTE: Based on pregnancy rates, this table compares the success rates for various time periods. The odds ratios (OR), 95% confidence intervals (CI), raw and adjusted p-values, as well as p-values for between-group comparisons, are displayed.
(Please view attached pdf to view all tables)
4. Discussion
Due to its adaptability in permitting the circumvention of laws restricting embryo culture and its potential to improve treatment options with a freeze-all indication, zygotes are kept via cryopreservation in IVF lab practice.
(Chen et al., 2003; Diedrich et al., 2020) say that 2PNs are less likely to be damaged than oocytes because they are better at taking cryoprotectants, releasing water, and withstanding osmotic pressure after freezing. Also, keeping 2PN for possible future cleavage stage transfers may be a good idea, since cleavage stage eggs are less likely to become vitrified than blastocysts (Zeng et al., 2018). Before PN breakdown can happen, the zygotes must be properly vitrified during the G2 phase of the cell cycle. However, the best time has never been found, and the effects of vitrification timing on clinical results have not been studied yet.
This study looks at how the time between vitrification and insemination affects the chance of getting pregnant in freeze-thaw IVF/ICSI rounds. We show that a specific date within the 17–21 hpi window for 2PN stage vitrifying had no effect on the chance of pregnancy when all important factors that affect pregnancy were taken into account. Keep in mind that in a single-factor trial, the chance of getting pregnant was more than double for embryo transfers with vitrification that happened between 19:01 and 21:00 hpi than for those that happened before 18:00 hpi.
We think that the difference between the results of the single and multivariate analyses is because the sample size was small and the study was done in the past. Based on our power analysis, it would be best to study the link between the amount of time between insemination and vitrification and the chance of getting pregnant with a bigger group.
Some of the confounding factors happen so rarely that a bigger sample might help the difficult multivariate analysis even more. A detailed look at the confounders in both the pregnant and non-pregnant study groups backs up this idea. Even well-known indicators of pregnancy risk, like the mother's age, AMH level, uterine pathology, and the amount of mature eggs, couldn't show a statistically significant link with pregnancy risk. Even though it wasn't important in the single study, the effect on the chance of getting pregnant was still there in the multivariate model.
According to the results of our study (Bushaqer et al., 2020), a successful embryo transfer in the past increased the chances of a good pregnancy test, which is in line with other research. Our single study also showed a strong link between the chance of getting pregnant and male factor infertility, which can be fixed with ICSI. Only 57 transfers were finished in Group C (19:01-21:00 hpi), which shows that changes between the groups of less than 20% did not have enough power and were unlikely to cause a 20% difference. The main problems with getting an accurate multifactorial analysis are the small sample size and the uneven spread of cases across the different time groups. Because our sample size was so small, we couldn't control for different ways of getting pregnant, like IVF vs. ICSI. The idea that, for example, the time of PNBD changes between IVF and ICSI morphokinetic traits would be a good reason to do a split study based on insemination method. The small difference in time between IVF and ICSI may be because sperm don't have instant access to the cytoplasm of the egg during IVF, but they do during ICSI (Bodri et al., 2015). But other studies (Karlikaya et al., 2022) show that the type of fertilization may not have a big effect on when the PNBD happens. This could be because, according to Bungum et al. (2006), traditional IVF only needs 30 seconds of gamete co-incubation to lead to fertilization and give clinical results that are similar to those of longer traditional gamete co-incubation. It's easy to think that sperm entry in IVF happens faster than expected and that the insemination times for IVF and ICSI don't vary much. If data from IVF and ICSI were merged and a single window for zygote freezing that took into account both techniques was established, normal laboratory procedures would also be made considerably easier.
Because all of the fertilization operations were carried out at the same university fertility facility using the same criteria for all patients, the study has the benefit of having high-quality data. The effects of any "learning effect" on the results were drastically decreased by just included a woman's initial thawing cycle over the duration of the trial.
Regardless of the results of the multivariate analysis, we are comfortable in the interpretation of our findings, which warn against vitrifying zygotes too soon in the second stage and suggest postponing at least 19 h after insemination. This is because the no confounder univariate evaluation and the multivariate analysis showed broad agreement with the body of research. This finding fits with the changes that happen to the zygote when it moves from the G1 phase of the cell cycle to the M phase. Due to differences between zygotes and tThis result matches the changes that happen to the zygote when it goes from the G1 phase of the cell cycle to the M phase. Because zygotes are formed in different ways, it is hard to tell when the G2 phase begins and ends. Time-lapse studies (Coticchio et al., 2018) show that 2PNs go up on average 6 hpi after ICSI and then go down 23 hpi, when the zygote enters the M stage of its life cycle. Literature (Miller and Goldberg, 1995; Macas et al., 1998; Damario et al., 1999; Dowling-Lacey et al., 2011) gives times for cryopreservation with IVF or ICSI that range from 16 to 23 hpi after slow freezing or vitrification. The S phase of the cell cycle starts around 9–10 hpi and lasts for 3–5 h, according to Balakier et al. During this time, a lot of DNA is made by the 2PNs. Between 20 and 22 hpi, when most zygotes are in the G2 phase, right before PN breaks down and chromosomes clump together, the method works well. At this time, it is not advised to freeze zygotes because they are fragile and easy to damage. A second study by Capmany et al. (1996) found that the S phase begins between 8 and 14 hpi and lasts between 2 and 4 h. Based on these numbers, the zygote's S phase could be over between 10 and 18 hours post-fertilization. Nagy et al. (1994), van Wissen et al. (1995), Capmany et al. (1996), and Neuber et al. (2003) all found that PN breaks down between 18 and 31 hpi. It's hard to know when vitrification should happen because there are so many different situations. Experiments have been done between 17 and 21 hpi, right after the fertilization check and before the PN breaks down. Our results show that the chance of getting pregnant is much higher if you vitrify zygotes at the 2PN stage before 18 hpi rather than after 19 hpi. This difference could be because some of the zygotes in Group A (17:00–18:00 hpi) turned into glass while they were still in the S phase. Even though timelapse technology has been made and used by other kinds of cells, it can't be used to see zygotes move from the S phase to the G2 phase (Sakaue-Sawano et al., 2008). We recommend vitrification until 19 hpi as a safe option to freezing the cell at a sensitive point until a time-lapse method can be used for human eggs. (Asami et al., 2022) New study shows that 2PN zygotes start transcription before 18 hpi. This shows how important the date we suggested is. Asami et al. have found that embryonic gene activation (EGA) begins after fertilization and continues until the embryo has 8 cells. The way they are fertilized, it is hard to know when the G2 phase starts and ends. Time-lapse studies (Coticchio et al., 2018) show that 2PNs rise on average 6 hpi after ICSI and then go down 23 hpi, when the zygote enters the M stage of its life cycle. After slow freezing or vitrification, the literature (Miller and Goldberg, 1995; Macas et al., 1998; Damario et al., 1999; Dowling-Lacey et al., 2011) lists times for cryopreservation with IVF or ICSI that range from 16 to 23 hpi. Balakier et al. (1993) say that the S phase of the cell cycle begins around 9–10 hpi and lasts for 3–5 h. During this phase, the 2PNs make a lot of DNA. When most zygotes are in the G2 phase between 20 and 22 hpi, right before PN breakdown and chromosomal condensation, the method performs well. Cryopreservation is never recommended at this time because zygotes are delicate and easily damaged. Capmany et al. (1996) did a second study and found that the S phase starts between 8 and 14 hpi and lasts between 2 and 4 h. Based on these figures, the S phase of the zygote could be over between 10 and 18 hpi. Nagy et al. (1994), van Wissen et al. (1995), Capmany et al. (1996), and Neuber et al. (2003) have all found that PN breaks down between 18 and 31 hpi. Because there is a lot of variety, it is hard to figure out when vitrification should happen. Experiments have been done between 17 and 21 hpi, right after the fertilization check and before PN breakdown. Our results show that the chance of getting pregnant is much higher if you vitrify zygotes at the 2PN stage before 18 hpi than if you do it after 19 hpi. This difference might be because some of the zygotes in Group A (17:00–18:00 hpi) turned into glass while they were still in the S phase. Even though other cell types have made and used timelapse technology, it can't be used to see the change from the S phase to the G2 phase in zygotes (Sakaue-Sawano et al., 2008). We suggest vitrification until 19 hpi as a safe alternative to freezing the cell at a sensitive stage until a time-lapse method can be used for human eggs. Recent research shows that 2PN zygotes start transcription before 18 hpi (Asami et al., 2022). This makes the date we suggested even more important. Research by Asami et al. has shown that embryonic gene activation (EGA) starts after fertilization and doesn't stop until the embryo has 8 cells. This is different from what was thought before. Scientists also found that some genes involved in cancer processes are among the first to be copied at the zygote. So, poor EGA, like that caused by vitrification, could be bad for the health of children. (Sargisian et al., 2022) There is more and more proof that both vitrification and frozen-thawed embryo transfer may increase the chance of cancer in children. Thus, the least invasive time to perform zygote vitrification may be considered a period when the delicate and special molecular machinery of zygotes is not hampered. To further understand the aforementioned idea, it would be intriguing to examine how transfers of thawed zygotes alter the outcomes of babies.
5. Conclusion
In conclusion, we found that delaying vitrification at the 2PN zygote stage to 19 HPI can increase the chances of pregnancy. Our results can help clarify the correct timing of vitrification and prevent the growth potential of transplanted embryos from being impaired. To assess the importance of the vitrification time in relation to relevant factors, we ask the community to replicate our results using big datasets. Maximum vitrification should be a key aim, in along with life and post-warm reproductive goals, in light of newly discovered findings linking frozen-thawed embryo transfer to pediatric cancer.
6. Future Directions
Future directions for research in the field of reproductive biology and assisted reproductive technologies may include the following areas:
Improving cryopreservation methods: Future study on cryopreservation methods, such as vitrification, could focus on improving processes to make it more likely that frozen-thawed embryos will live, be healthy, and be able to grow and develop. This includes looking into new cryoprotectants and methods for freezing and thawing, as well as figuring out what the long-term effects of cryopreservation are on the growth of embryos and the health of their offspring.
Embryo selection and viability prediction: To improve pregnancy rates and cut down on multiple pregnancies, it is important to find more accurate ways to choose embryos and predict their chances of survival. Advanced technologies like time-lapse images, metabolomic profiling, genetic testing, and artificial intelligence programs could be used to test the quality of embryos and predict how likely they are to implant.
Conditions for growing embryos: It is important to find out what the best conditions are for growing embryos in the lab in order to help them grow and improve their chances of success. Researchers could try to find the best culture medium, oxygen levels, and other external factors that mimic the conditions in vivo. They could also look at the effects of long-term culture on embryo growth and the chance that it will implant.
Single embryo transfer (SET) and fewer multiple pregnancies: The goal of assisted reproductive technologies should be to reach SET while keeping success rates high. More study could look into ways to improve the rate of embryo selection and implantation, making SET safer and more successful and lowering the risks of having more than one baby.
Long-term health effects: It is very important to study the long-term health effects of children who were born with the help of assisted reproductive technologies. Researchers could study the effects of cryopreservation, hormonal stimulation, and cell growth on the health and development of children throughout their lives.
Personalized approaches: Tailoring fertility treatments to individual patients based on their specific characteristics and needs could improve outcomes. Future research could investigate the use of personalized medicine approaches, including genetic profiling, phenotypic assessment, and individualized treatment protocols, to optimize fertility treatments and increase success rates.
Ethical and social considerations: As assisted reproductive technologies advance, there is a need for ongoing research on the ethical, legal, and social implications of these technologies. Studies could explore the impact of fertility treatments on individuals, families, and society, addressing issues such as access to care, equity, informed consent, and the psychological well-being of patients and offspring.
These are just a few potential areas for future research in reproductive biology and assisted reproductive technologies. Continued scientific investigation and innovation in these areas can contribute to improving fertility treatments, enhancing success rates, and ensuring the well-being of individuals and families undergoing assisted reproduction.
References
1.Al-Hasani, S., Ozmen, B., Koutlaki, N., Schoepper, B., Diedrich, K., and Schultze-Mosgau, A. (2007). Three years of routine vitrification of human zygotes: Is it still fair to advocate slow-rate freezing? Reprod. Biomed. Online 14, 288–293. doi:10.1016/s1472-6483(10)60869-3
2.Asami, M., Lam, B. Y. H., Ma, M. K., Rainbow, K., Braun, S., VerMilyea, M. D., et al. (2022). Human embryonic genome activation initiates at the one-cell stage. Cell Stem Cell 29, 209–216.e4. doi:10.1016/j.stem.2021.11.012
3.Azzarello, A., Hoest, T., and Mikkelsen, A. L. (2012). The impact of pronuclei morphology and dynamicity on live birth outcome after time-lapse culture. Hum. Reprod. 27, 2649–2657. doi:10.1093/humrep/des210
4.Balakier, H., MacLusky, N. J., and Casper, R. F. (1993). Characterization of the first cell cycle in human zygotes: Implications for cryopreservation. Fertil. Steril. 59, 359–365. doi:10.1016/s0015-0282(16)55678-7
5.Bodri, D., Sugimoto, T., Serna, J. Y., Kondo, M., Kato, R., Kawachiya, S., et al. (2015). Influence of different oocyte insemination techniques on early and late morphokinetic parameters: Retrospective analysis of 500 time-lapse monitored blastocysts. Fertil. Steril. 104 (5), 1175–1181.e812. doi:10.1016/j.fertnstert.2015.07.1164
6.Bungum, M., Bungum, L., and Humaidan, P. A. (2006). A prospective study, using sibling oocytes, examining the effect of 30 seconds versus 90 minutes gamete co-incubation in IVF. Hum. Reprod. 21 (2), 518–523. doi:10.1093/humrep/dei350
7.Bushaqer, N. J., Alkhudhairy, N. N., Alturaigi, Z. M., Alhamad, R. M., Mohawesh, W. A., Alraka, F. E., et al. (2020). The effect of fresh IVF cycle characteristics on frozen embryo transfer (FET) outcomes. JBRA Assist. Reprod. 24, 135–142. doi:10.5935/1518-0557.20190074
8.Capmany, G., Taylor, A., Braude, P. R., and Bolton, V. N. (1996). The timing of pronuclear formation, DNA synthesis and cleavage in the human 1-cell embryo. Mol. Hum. Reprod. 2, 299–306. doi:10.1093/molehr/2.5.299
9.Chen, S. U., Lien, Y. R., Chao, K. H., Ho, H. N., Yang, Y. S., and Lee, T. Y. (2003). Effects of cryopreservation on meiotic spindles of oocytes and its dynamics after thawing: Clinical implications in oocyte freezing - a review article. Mol. Cell. Endocrinol. 202, 101–107. doi:10.1016/s0303-7207(03)00070-4
10.Chen, Y., Nisenblat, V., Yang, P., Zhang, X., and Ma, C. (2018). Reproductive outcomes in women with unicornuate uterus undergoing in vitro fertilization: A nested case-control retrospective study. Reprod. Biol. Endocrinol. 16, 64. doi:10.1186/s12958-018-0382-6
11.Coccia, M. E., Rizzello, F., Wakunga, S., Badolato, L., Evangelisti, P., Bertocci, F., et al. (2020). Two countries-two labs: The transnational gamete donation (TGD) programme to support egg donation. J. Assist. Reprod. Genet. 37, 3039–3049. doi:10.1007/s10815-020-01961-w.
12.Cohen, J. (1998). Statistical power analysis for the behavioral science. (N. Y. U. Department of Psychology New York: Lawrence Erlbaum Associates).
13.Coticchio, G., Mignini Renzini, M., Novara, P. V., Lain, M., De Ponti, E., Turchi, D., et al. (2018). Focused time-lapse analysis reveals novel aspects of human fertilization and suggests new parameters of embryo viability. Hum. Reprod. 33, 23–31. doi:10.1093/humrep/dex344
14.Damario, M. A., Hammitt, D. G., Galanits, T. M., Session, D. R., and Dumesic, D. A. (1999). Pronuclear stage cryopreservation after intracytoplasmic sperm injection and conventional IVF: Implications for timing of the freeze. Fertil. Steril. 72, 1049–1054. doi:10.1016/s0015-0282(99)00444-6
15.De Munck, N., Bayram, A., Elkhatib, I., Abdala, A., El-Damen, A., Arnanz, A., et al. (2022). Marginal differences in preimplantation morphokinetics between conventional IVF and ICSI in patients with preimplantation genetic testing for aneuploidy (PGT-A): A sibling oocyte study. PloS One 17 (4), e0267241. doi:10.1371/journal.pone.0267241
16.Diedrich, K., Ludwig, M., and Ludwig, M. (2020). Reproduktionsmedizin. 2., erweiterte und vollständig überarbeitete. Deutschl: Springer Ref. Medizin.
17.Fancsovits, P., Toth, L., Takacs, Z. F., Murber, A., Papp, Z., and Urbancsek, J. (2005). Early pronuclear breakdown is a good indicator of embryo quality and viability. Fertil. Steril. 84, 881–887. doi:10.1016/j.fertnstert.2005.03.068
18.Gallego, R. D., Remohí, J., and Meseguer, M. (2019). Time-lapse imaging: The state of the art. Biol. Reprod. 101, 1146–1154. doi:10.1093/biolre/ioz035
19.Golakov, M., Depenbusch, M., Schultze-Mosgau, A., Schoepper, B., Hajek, J., Neumann, K., et al. (2018). What is the net effect of introducing vitrification for cryopreservation of surplus 2PN oocytes in an IVF program? Arch. Gynecol. Obstet. 297, 529–537. doi:10.1007/s00404-017-4606-3
20.Huo, Y., Yuan, P., Qin, Q., Yan, Z., Yan, L., Liu, P., et al. (2021). Effects of vitrification and cryostorage duration on single-cell RNA-Seq profiling of vitrified-thawed human metaphase II oocytes. Front. Med. 15, 144–154. doi:10.1007/s11684-020-0792-7
21.Karlikaya, G., F?nd?kl?, N., Aksoy, T., Olcay, O., Teke, B., Boynukal?n, K., et al. (2022). Farkl? inseminasyon teknikleri ile elde edilmi? insan embriyolar?n?n erken fertilizasyon ve klivaj dinamikleri yönünden morfokinetik analizi: Prospektif karde? oosit çal??mas?. Jinekoloji-Obstetrik ve Neonatoloji T?p Derg. 19 (1), 1135–1141. doi:10.38136/jgon.1031669
22.Kuwayama, M. (2007). Highly efficient vitrification for cryopreservation of human oocytes and embryos: The cryotop method. Theriogenology 67, 73–80. doi:10.1016/j.theriogenology.2006.09.014
23.Macas, E., Imthurn, B., Borsos, M., Rosselli, M., Maurer-Major, E., and Keller, P. J. (1998). Impairment of the developmental potential of frozen-thawed human zygotes obtained after intracytoplasmic sperm injection. Fertil. Steril. 69, 630–635. doi:10.1016/s0015-0282(98)00021-1
24.Miller, K. F., and Goldberg, J. M. (1995). In vitro development and implantation rates of fresh and cryopreserved sibling zygotes. Obstet. Gynecol. 85, 999–1002. doi:10.1016/0029-7844(95)00084-5
25.Montag, M., van der Ven, K., Dorn, C., and van der Ven, H. (2006). Extended embryo culture reduces the implantation rate on day 4 and day 5 when only a maximum of three embryos are cultured beyond the pronuclear stage. Eur. J. Obstet. Gynecol. Reprod. Biol. 124, 65–69. doi:10.1016/j.ejogrb.2005.08.022
26.Nagy, Z. P., Liu, J., Joris, H., Devroey, P., and Van Steirteghem, A. (1994). Time-course of oocyte activation, pronucleus formation and cleavage in human oocytes fertilized by intracytoplasmic sperm injection. Hum. Reprod. 9, 1743–1748. doi:10.1093/oxfordjournals.humrep.a138786
27.Neuber, E., Rinaudo, P., Trimarchi, J. R., and Sakkas, D. (2003). Sequential assessment of individually cultured human embryos as an indicator of subsequent good quality blastocyst development. Hum. Reprod. 18, 1307–1312. doi:10.1093/humrep/deg269
28.Nikolettos, N., and Al-Hasani, S. (2000). Frozen pronuclear oocytes: Advantages for the patient. Mol. Cell. Endocrinol. 169, 55–62. doi:10.1016/s0303-7207(00)00352-x
29.Rienzi, L., Gracia, C., Maggiulli, R., LaBarbera, A. R., Kaser, D. J., Ubaldi, F. M., et al. (2017). Oocyte, embryo and blastocyst cryopreservation in ART: Systematic review and meta-analysis comparing slow-freezing versus vitrification to produce evidence for the development of global guidance. Hum. Reprod. Update 23, 139–155. doi:10.1093/humupd/dmw038
30.Sakaue-Sawano, A., Kurokawa, H., Morimura, T., Hanyu, A., Hama, H., Osawa, H., et al. (2008). Visualizing spatiotemporal dynamics of multicellular cell-cycle progression. Cell 132, 487–498. doi:10.1016/j.cell.2007.12.033
31.Sargisian, N., Lannering, B., Petzold, M., Opdahl, S., Gissler, M., Pinborg, A., et al. (2022). Cancer in children born after frozen-thawed embryo transfer: A cohort study. PLoS Med. 19, e1004078. doi:10.1371/journal.pmed.1004078
32.Senn, A., Vozzi, C., Chanson, A., De Grandi, P., Germond, M., and Chanson, A. (2000). Prospective randomized study of two cryopreservation policies avoiding embryo selection: The pronucleate stage leads to a higher cumulative delivery rate than the early cleavage stage. Fertil. Steril. Nov. 74 (5), 946–952. doi:10.1016/s0015-0282(00)01603-4
33.Shapiro, B. S., Daneshmand, S. T., Garner, F. C., Aguirre, M., and Hudson, C. (2015). Freeze-all at the blastocyst or bipronuclear stage: A randomized clinical trial. Fertil. Steril. 104, 1138–1144. doi:10.1016/j.fertnstert.2015.07.1141
34.Surrey, E. S., Minjarez, D. A., Stevens, J. M., and Schoolcraft, W. B. (2005). Effect of myomectomy on the outcome of assisted reproductive technologies. Fertil. Steril. 83, 1473–1479. doi:10.1016/j.fertnstert.2004.11.045
35.Troup, S. A., Matson, P. L., Critchlow, J. D., Morroll, D. R., Lieberman, B. A., and Burslem, R. W. (1991). Cryopreservation of human embryos at the pronucleate, early cleavage, or expanded blastocyst stages. Eur. J. Obstet. Gynecol. Reprod. Biol. 38 (2), 133–139. doi:10.1016/0028-2243(91)90190-v