Myasthenia Gravis (MG): An In-depth Approach A Case Report of Myasthenia Gravis after Forty Years of Neurology Work
Myasthenia Gravis (MG): An In-depth Approach
A Case Report of Myasthenia Gravis after Forty Years of Neurology Work
Dr. Hassan Jazayeri *
*Correspondence to: Dr. Hassan Jazayeri. Neuropsychologist.
Copyright
© 2025 Dr. Hassan Jazayeri, This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 05 Aug 2025
Published: 28 Aug 2025
DOI: https://doi.org/10.5281/zenodo.17043159
Abstract
A patient who, with symptoms of shortness of breath and difficulty swallowing, referred to the emergency room and was hospitalized in the neurology department, was finally diagnosed with Myasthenia gravis. This diagnosis was confirmed based on the positive results of some tests. Although the thymus gland was reported to be normal and the patient's condition was almost stabilized with the classic treatment of Myasthenia.
With the patient's weight gain, due to the use of corticosteroids, although his family and nutritionist insisted on taking the patient to strict diets and even 12 or 16-hour fasts, we unilaterally opposed these requests. Our point was that:
We should not put any additional stress on the patient at this stage. Because would complicate the problem. Currently, the patient is undergoing a natural process of treatment and control.
Myasthenia Gravis (MG): An In-depth Approach A Case Report of Myasthenia Gravis after Forty Years of Neurology Work
Introduction
Myasthenia Gravis (MG) is a rare, chronic autoimmune disease, characterized by fluctuating muscle weakness and fatigue. It is caused by a disorder of neuromuscular transmission at the neuromuscular junction (NMJ). Given the complexities of its pathogenesis, the wide range of clinical manifestations, and recent advances in therapeutic approaches, a thorough understanding of this disease is essential for neurologists. The aim of this article is to provide a comprehensive and up-to-date review of key aspects of MG, focusing on the specialized information needed for the clinical management of patients.
First Case:
The first time, I introduced to myasthenia gravis, at 1971, before I started Medical Faculty. I still remember when I saw my friend's drooping eyelid, and then both of them. I kept reminding him that this drooping eyelid was abnormal. But he said, "I don't have any problem." It causes problem, when the weather is hot. The city where we lived was relatively cold. Later, he went to Turkey and studied there and had swallowing problems, which eventually led to him being treated by a neurologist.
After this case, I don't remember seeing a case of Myasthenia gravis. Almost 63 years later, one day, my colleagues who work with me, asked to visit an interesting case. When I met this 77-year-old man, his right eye ptosis was so obvious that I asked him:
- How long have you had this problem?
He said:
- Three months.
But he immediately emphasized that his main problem was difficulty swallowing food. He emphasized that food goes down with difficulty. For this reason, I tend to drink liquids. Although drinking fluids is accompanied by its jumping into the airways, which makes me feel suffocated.
The man told me:
- Along with these problems, weakness and lethargy are very pressing.
When I put all the patient's problems together, I focused on Myasthenia gravis and decided to rule it. Thus, this case started.
Pathogenesis and Immunopathogenesis:
The pathogenesis of MG is mainly based on the production of autoantibodies against specific proteins in the NMJ. The most common antibody is against nicotinic acetylcholine receptors (AChR) in the postsynaptic membrane, which is found in about 85% of AChR-MG positive patients. These antibodies lead to disruption of neuromuscular transmission through various mechanisms:
• Receptor destruction:
Antibodies can bind to AChRs, causing their endocytosis and destruction, which leads to a decrease in the number of active receptors.
• Acetylcholine binding site blocking:
Antibodies can competitively block the acetylcholine binding site and prevent the binding of the neurotransmitter to the receptors.
• Complement-mediated destruction:
Activation of the complement system by antibody-AChR immune complexes leads to damage and destruction of the postsynaptic membrane.
Fig 1
Other types of antibodies:
• Antibody against muscle-specific kinase (MuSK-Ab):
Antibodies against MuSK are found in about 5-10% of MG patients, especially in patients without AChR antibodies (serum-negative). MuSK is a tyrosine kinase at the NMJ that is involved in the formation and maintenance of AChR clusters. MuSK-MG patients often have a different clinical phenotype, including more prominent involvement of the swallowing, respiratory, and neck muscles.
• Antibody to lipoprotein receptor-complement 4 (LRP4-Ab):
LRP4 acts as an agonist for MuSK and is involved in AChR clustering. Antibodies to LRP4 are found in a small subset of seronegative patients.
• Antibodies to titin (Titin-Ab) and ranodyne receptor (RyR-Ab):
These antibodies are more commonly seen in MG patients with thymoma and/or late-onset MG.
Fig 2
• Other emerging antibodies:
Research is identifying new antibodies that may be involved in the pathogenesis of MG, such as antibodies to other unknown NMJ proteins.
Role of the thymus: The thymus plays a central role in the pathogenesis of MG. About 10-15% of MG patients have thymomas (tumors of the thymus) and 70% have thymic hyperplasia. The thymus is known to be the main site of production of autoreactive T cells and synthesis of AChR antibodies.
Prevalence and epidemiology:
MG is a rare disease, but its prevalence is increasing.
• Prevalence: approximately 150-250 people per million.
• Incidence: approximately 10-20 new cases per million per year.
• Gender:
Women: more often affected at a young age (20s and 30s).
Men: more often affected at an older age (50s and 60s).
• Race and communities:
MG occurs in all races and ethnic groups.Ethnicity is seen. Recent studies have shown that its prevalence may be slightly higher in some Asian populations, especially in association with certain types of antibodies (such as MuSK-MG).
Clinical features:
MG is characterized by fluctuating muscle weakness and fatigue that worsens with activity and improves with rest. Symptoms can range from mild to severe and life-threatening:
• Ocular weakness:
The most common initial manifestation (about 50-60% of patients). Includes unilateral or bilateral ptosis (drooping eyelids) and diplopia (double vision) due to involvement of extraocular muscles.
• Bulbar weakness:
Involvement of the pharynx and larynx muscles leads to dysphagia (difficulty swallowing), dysphonia (difficulty speaking), and hoarseness. These manifestations can increase the risk of aspiration.
• Limb weakness:
Proximal muscle weakness (more common than distal. Upper limbs are often more commonly affected than lower limbs.
• Respiratory weakness:
Weakness of the diaphragm and intercostal muscles that can lead to respiratory failure and myasthenic crisis; considered a medical emergency.
• Neck weakness:
Weakness of the neck muscles that can lead to head drop.
• Disease course:
About 50% of patients with ocular MG progress to diffuse MG within two years. MuSK-MG patients are more likely to have more prominent swallowing and respiratory involvement.
Diagnosis:
Diagnosis of MG requires a multifaceted approach that includes clinical evaluation, Laboratory and electrophysiological:
1. Clinical examination and neurological examination:
- Careful assessment of muscle strength, fatigue, and reflexes.
- Weakness exacerbation tests: such as gaze-up (for ptosis), one-breath counting (for dysphagia/respiratory weakness), or arm muscle fatigue test.
2. Laboratory tests:
- Serum antibodies:
Anti-AChR antibody: The most important diagnostic test with a sensitivity of 85-90% in diffuse MG and 50-70% in ocular MG.
Anti-MuSK antibody: Should be requested in AChR-Ab negative patients with high clinical suspicion of MG.
Anti-LRP4 antibody: Currently not recommended for routine diagnosis but may be useful in sero-negative cases refractory to diagnosis.
- Related screening tests: Thyroid function test (due to higher prevalence of autoimmune thyroid diseases in MG) and screening for other autoimmune diseases Related.
3. Electrophysiological studies:
- Repetitive Nerve Stimulation (RNS): Looks for a >10% decrease in the amplitude of the compound muscle action potential (CMAP) after repeated nerve stimulation (2-3 Hz). The sensitivity of RNS is about 70-80% in diffuse MG and less in ocular MG.
- Single Fiber Electromyography (SFEMG): The most sensitive electrophysiological test for MG (sensitivity >90% in diffuse MG and 95% in ocular MG). Looks for increased jitter (impaired timing of action potentials of two muscle fibers) and block (absence of action potential in one of the fibers). SFEMG is recommended in patients with a negative RNS and high clinical suspicion of MG.
4. Imaging:
- Chest CT scan or MRI: To evaluate the thymus and look for thymoma or thymic hyperplasia. This part of the diagnosis is essential for all patients with MG.
Differential diagnoses:
This disease It should be differentiated from other NMJ disorders (e.g. Lambert-Eaton syndrome, botulism), other neuromuscular diseases (e.g. amyotrophic lateral sclerosis, myopathies), and non-neurological causes of muscle weakness. The reality is that in poor countries and lack of access to extensive diagnostic facilities, we cannot practically use all the diagnostic capabilities for rare diseases such as myasthenia gravis. Many cases and the financial situation of the patients do not allow such a start for diagnosis and until the patient has reached a critical condition, it may not be practical to hospitalize the patient in government departments for diagnosis alone. For this reason, we have to start treatment with some positive tests and monitor the disease and determine the outlook based on the progress of treatment and response to treatment. This disease is an emergency at night, because a piece of food has fallen into the bronchial area while eating and the patient's attempts to expel the food with repeated coughing have not led to anything. When the patient was admitted to the hospital as an emergency, he was admitted to the emergency department to resolve the patient's problem and then transferred to the neurology department. The suspicion of myasthenia gravis was started.
Treatment:
The goal of MG treatment is to control symptoms, prevent disease exacerbation, and improve the patient's quality of life. The therapeutic approach is determined based on the severity of the disease, the presence of thymoma, and the patient's response to previous treatments:
1. Symptomatic treatments:
- Acetylcholinesterase inhibitors (AChEIs):
Pyridostigmine is the most common drug in this class. By inhibiting the acetylcholinesterase enzyme, the concentration of acetylcholine at the NMJ increases and neuromuscular transmission improves.
2. Immunomodulatory treatments:
- Corticosteroids:
Prednisone is the first line of immunomodulatory treatment. They work by suppressing the immune system.
- Nonsteroidal immunosuppressive drugs:
To reduce the dose of steroids or in cases Resistant to treatment. Includes Azathioprine, Mycophenolate Mofetil, Cyclosporine, and Tacrolimus.
3. Rapid-acting treatments in acute cases:
- Plasma Exchange (PLEX):
To rapidly remove antibodies from the circulation. In myasthenic crisis, before Thymectomy surgery, or in cases of severe exacerbations.
- Intravenous Immunoglobulin (IVIg):
Used in myasthenic crisis and in cases of acute exacerbations.
4. Thymectomy:
- Indications:
Recommended in all MG patients with thymoma. In patients 18 to 65 years of age without thymoma and with positive AChR antibodies, thymectomy may result in clinical improvement or reduced need for immunosuppressive drugs.
We considered thymoma a lot when examining the patient's thymus, especially since it had progressed so rapidly, but in the end, not only did we not find it, but the size of the thymus was also found to be very small and in fact had no effect on the symptoms of myasthenia gravis in this patient.
New and targeted therapies:
Monoclonal antibodies:
- Rituximab:
An anti-CD20 antibody that targets B cells. It is particularly effective in patients with refractory MuSK-MG.
- Eculizumab and rovelizumab:
C5 complement inhibitors, approved for refractory disseminated MG in AChR-MG-positive patients.
- Efgartigimod:
A neonatal Fc receptor (FcRn) antagonist that reduces levels of pathogenic antibodies.
Pregnancy and MG
Management of MG during pregnancy requires a careful, multidisciplinary approach.
• Changes in disease severity:
The severity of MG can fluctuate during pregnancy; Some women recover and others experience exacerbations. The first trimester and postpartum period are considered high-risk periods for exacerbations.
• Medications:
- Pyridostigmine:
Safe and can be used in pregnancy.
- Prednisone:
Usually safe in pregnancy, but monitoring for side effects is required.
- Azathioprine:
Can be used with caution if necessary.
- Mycophenolate mofetil:
Absolutely contraindicated in pregnancy due to teratogenicity.
- PLEX and IVIg:
Safe in pregnancy and can be used in cases of crisis or severe exacerbation.
• Delivery:
Normal delivery is usually possible for MG patients. Analgesic epidural is preferred. Close monitoring of respiratory function and fatigue management are required during delivery. Cesarean section is performed only if obstetrically indicated.
• Neonates:
Transient Neonatal Myasthenia Gravis (TNMG) is seen in 10-20% of infants born to MG-positive mothers, resulting from the transfer of maternal antibodies from the placenta to the fetus. This condition is usually self-limiting and transient, resolving within a few weeks or months.
Challenges and Future Prospects
Despite significant advances in the understanding and management of MG, several challenges remain:
• Sero-negative MG:
There is still a subgroup of patients who lack known antibodies, and their diagnosis and treatment are more complex. Identification of novel antibodies and diagnostic biomarkers is essential.
• Predicting response to treatment:
The lack of clinical or biological predictors of response to treatment makes the selection of an appropriate treatment strategy challenging.
• Optimization of existing treatments:
The need to find more effective and less toxic doses of immunosuppressive drugs and reduce long-term side effects.
• New targeted therapies:
Development of drugs that more specifically target the pathogenic pathways of MG (such as selective inhibitors of T or B cells, or molecules involved in neuromuscular interaction).
• Animal and cellular models:
Development of more clinically relevant models to study pathogenesis and evaluate new drugs.
• Quality of life:
Comprehensive management of MG patients also includes aspects of rehabilitation, psychological support, and improvement of their overall quality of life.
• Dementia:
Despite the fact that the mother of the studies of other cases of myasthenia gravis did not find cases of dementia or memory disorders, and since myasthenia itself cannot cause dementia or even Alzheimer's, we did not have access to a reliable document. However, in our case, which we closely follow the issue and are doing, we were able to find effects of senile dementia. And when we also performed the MMSE test, we surprisingly found a complete and significant drop in this test, which although is still far from serious dementia disorders and especially Alzheimer's, this concern for us and those around the patient still exists and we cannot ignore this issue. Especially since before this issue, the patient had not raised any doubts for us as a fetus. While now, if we cannot control the patient, we are forced to use a caregiver.
Prognosis
The prognosis of MG has improved significantly with recent therapeutic advances. The majority of patients experience significant improvement in symptoms and quality of life with appropriate management. However:
• Complete remission: Occurs in a small number of patients (especially after thymectomy in some cases).
• Exacerbations: May occur throughout the course of the disease, especially during periods of stress, infection, or nonadherence to treatment.
• Myasthenic crisis: Remains a serious and life-threatening complication that requires emergency management.
Dos and don'ts in managing MG
Dos:
• Patient education:
Educate patients about the disease, symptoms of exacerbations, and the importance of adherence to treatment.
• Close monitoring:
Monitor clinical symptoms, response to treatment, and adverse effects of medications regularly.
• Thymoma screening:
Perform chest CT or MRI for all MG patients.
• Vaccination:
Vaccinate patients to protect against infections, especially if taking immunosuppressive medications
• Tim Multidisciplinary:
Involve pulmonologists, gastroenterologists, and thoracic surgeons as needed.
• Psychological support:
Provide counseling and psychological support to patients and their families.
Do not:
• Contraindicated medications:
Avoid prescribing medications that can exacerbate MG (such as some antibiotics, beta-blockers, some sedatives).
• Abrupt discontinuation of treatment:
Especially corticosteroids or immunosuppressive drugs
• Ignoring respiratory or swallowing symptoms:
These symptoms may indicate the onset of a myasthenic crisis and require immediate action.
Conclusions
Myasthenia gravis is a complex and multifaceted disease, the management of which requires a deep understanding of its pathogenesis and the wide range of its clinical manifestations. With recent advances in immunology and pharmacology, life expectancy and quality of life in MG patients have improved significantly. However, further research is necessary to discover new treatments, optimize existing approaches, and ultimately find a definitive cure for this disease. Collaboration and collaboration between neurologists, immunologists, surgeons, and other disciplines is crucial to provide the best possible care for MG patients.
Our patient was admitted to the neurology department three times in a four-month period as an emergency, including for dysphagia, dyspnea, and nausea and vomiting, and a thorough clinical examination revealed that he also suffered from speech problems, diplopia, and bilateral drooping of the eyes. . Finally, with the measures taken in the public sector, we have reached the diagnosis. That is, myasthenia gravis grade IIIA according to the MGFA classification, positive for AchR antibodies
Even for immunological tests, the patient's samples were sent to three countries: Greece, Italy, and Germany, because such capacities for such tests do not currently exist in Albania itself. At the same time, during the same four-month period, the patient had herpes, which has improved with anti-herpes treatments. The treatments used for this patient were prednisolone tablets, mestinon tablets, azathioprine.
Interestingly, the gastroscopy report of the patient reported severe candidiasis in the esophagus and recommended oral treatment with nystatin. The EMG and ENG tests performed on the patient confirmed myasthenia gravis. In future studies, the issue of dementia or Alzheimer's should be investigated, especially in cases where the patient is elderly.
Unilateral treatment:
In this case, when the patient was treated with prednisolone × at a very high dose for a while, some of the patient's family, and especially one of our colleagues who follows up on nutrition for patients, insisted that, considering the patient's weight gain and the patient's wife ×, he should definitely be placed on a restricted diet program and even a 12- or 16-hour fast. I strongly opposed this plan. My point was that we should not expose a patient with these symptoms to other challenges that, apart from increasing stress and thus slowing down the patient's recovery process, will not solve the serious problem for him. When the patient's condition gradually reaches a stable state, the patient's weight will definitely decrease with a decrease in the dose of prednisolone. Even psychologically, raising this issue with the patient can create non-organic reflections in the patient that we did not want.
References
1. Gilhus, N. E. (2019). Myasthenia Gravis. New England Journal of Medicine, 381(19), 1861-1871.
2. Sanders, D. B., Wolfe, G. I., Burns, T. M., & Massey, J. M. (2016). Myasthenia gravis. Seminars in Neurology, 36(5), 415-429.
3. Skeie, G. O., & Evoli, A. (2021). The diagnosis and management of myasthenia gravis in Europe: a consensus statement. European Journal of Neurology, 28(6), 1640-1651.
4. Farmakidis, C., Souayah, N., & Brannagan III, T. H. (2018). Myasthenia gravis: an update on diagnosis and management. Journal of Clinical Neuromuscular Disease, 19(2), 53-61.
5. Sanders, D. B., Wolfe, G. I., Burns, T. M., & Massey, J. M. (2016). Myasthenia gravis. Seminars in Neurology, 36(5), 415-429.
6. Murthy, J. M., Singh, S. K., & Pradhan, S. (2018). Myasthenia gravis: a comprehensive review. Annals of Indian Academy of Neurology, 21(5), S1-S12.
7. Koli, D., Taneja, R., Yadav, V., & Das, S. (2023). Novel therapeutics for myasthenia gravis: from bench to bedside. Immunopharmacology and Immunotoxicology, 45(1), 1-13.
8. Svensson, B. A., & Thorlacius, L. (2020). Epidemiology of myasthenia gravis. Journal of Neuromuscular Diseases, 7(2), 117-123.
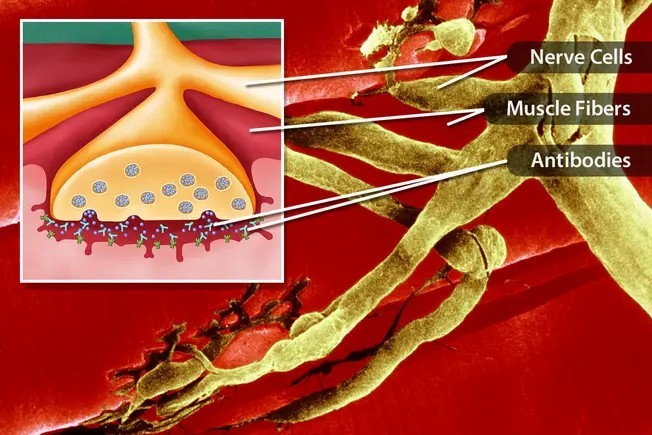
Figure 1
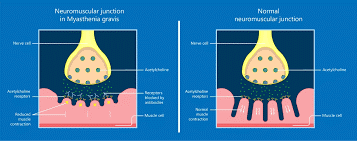
Figure 2