Pancreatic Neuroendocrine Tumor Associated with Tuberous Sclerosis Complex: A Case Report
Pancreatic Neuroendocrine Tumor Associated with Tuberous Sclerosis Complex: A Case Report
Arenas T1, Gordienko B1, Agüero P1, Pineyro Maria M1, Del Pozo E1*
1. Academic Unit of Endocrinology and Metabolism, Hospital de Clínicas, Faculty of Medicine, University of the Republic, Montevideo, Uruguay.
Del Pozo E: https://orcid.org/0009-0003-2215-8378.
*Correspondence to: Del Pozo E, Academic Unit of Endocrinology and Metabolism, Hospital de Clínicas, Faculty of Medicine, University of the Republic, Montevideo, Uruguay.
Copyright.
© 2025 Arenas T This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 19 Aug 2025
Published: 29 Aug 2025
DOI: https://doi.org/10.5281/zenodo.17038763
Abstract
Pancreatic neuroendocrine tumors (PNETs) are rare. They can be sporadic or associated with hereditary syndromes. The tuberous sclerosis complex (TSC) is associated with the occurrence of PNETs in about 1% of cases. Diagnosis of the tumor is made with biochemical markers and imaging techniques. Management in these cases remains controversial. We report a case of a 46-year-old male patient with a prior diagnosis of TSC and imaging findings of a pancreatic tumor. Clinical suspicion is essential in patients with predisposing conditions and requires evaluation by a multidisciplinary team.
Keywords: Pancreatic neuroendocrine tumor; tuberous sclerosis complex, genetic disorder.
Pancreatic Neuroendocrine Tumor Associated with Tuberous Sclerosis Complex: A Case Report
Introduction
Pancreatic neuroendocrine tumors (PNETs) are rare [1, 2] neoplasms with an incidence of ≤ 1 case per 100,000 individuals per year, being more common in females. Most PNETs are sporadic, but they may be associated with hereditary syndromes such as multiple endocrine neoplasia type 1 (MEN1), Von Hippel-Lindau disease (VHL), neurofibromatosis type 1 (NF1), and tuberous sclerosis complex (TSC) [4].
TSC is a rare autosomal dominant genetic disorder caused by mutations in TSC1 or TSC2, leading to multiple tumors in the skin, brain, kidneys, and lungs. Approximately 1% of TSC cases are associated with PNETs [5].
Case Report
We present the case of a 46-year-old male patient with a personal history of TSC. He had secondary epilepsy since the age of 4, treated with carbamazepine and valproate, moderate to severe intellectual disability, and bilateral renal angiomyolipomas. No relevant family history was reported.
The patient was referred by the General Surgery Department of the Hospital de Clínicas due to the incidental finding of a pancreatic tumor. The patient denied symptoms suggestive of functionality, metastatic disease, or general compromise.
Figure 1: Abdominal MRI. Red arrow: Exophytic tumor in the posterior sector of the pancreatic body.
Physical examination revealed overweight (BMI 29 kg/m2) and an unremarkable abdominal exam.
Initial laboratory tests: Glucose 91 mg/dL, Na 143 mEq/L, K 4.3 mEq/L, Cl 106 mEq/L, creatinine 0.78 mg/dL, urea 0.35 g/L, P 2.8 mg/dL, calcium 9.4 mg/dL, Hb 14.3 g/dL, WBC 5,330/mm³, platelets 138,000/mm³, gastrin 17.1 pg/mL (normal range 13-115), chromogranin A 61 ng/mL (normal <93).
MRI showed a pancreas of normal size and morphology. In the posterior body of the pancreas, a 30 mm solid exophytic nodule was observed, with diffusion restriction and intense arterial phase enhancement persisting in late phases. Wirsung duct was normal. (Figure 1)
PET/CT with Ga-DOTATATE revealed an 11 mm hypercaptant nodule in the pancreatic head and a known 30 mm lesion in the body. The case was discussed with the surgical team. Endoscopic biopsy was performed and treatment with octreotide 20 mg/day s.c. was initiated.
Pathology of the body lesion revealed no malignancy and normal pancreatic parenchyma. The head lesion showed neuroendocrine morphology without pleomorphism, anaplasia, or mitoses, consistent with a well-differentiated neuroendocrine tumor. Ki-67 could not be assessed.
The patient remains asymptomatic. The case was re-discussed in a multidisciplinary meeting, and a watchful waiting approach was maintained.
Discussion
PNETs are classified as functioning or non-functioning tumors, depending on the pancreatic endocrine cell type of origin and hormone secretion (insulinoma, gastrinoma, glucagonoma, VIPoma, somatostatinoma) [2, 6]. Non-functioning PNETs represent 10–50% of all PNETs and may present with local mass effects such as abdominal pain, palpable mass, jaundice, weight loss, or general deterioration, without hormone hypersecretion symptoms, as in our case [5, 6].
Biochemical diagnosis relies mainly on chromogranin A, although other markers such as pancreatic polypeptide or neuron-specific enolase can be measured [6]. Imaging modalities include CT, MRI, endoscopic ultrasound, somatostatin receptor scintigraphy (Octreoscan®), or PET, as used in this patient. Definitive diagnosis is histological [6].
Somatostatin analogs are indicated in several neuroendocrine tumors, mainly functioning ones to control symptoms, but they also show antiproliferative effects, improving progression-free survival in both pancreatic and extra-pancreatic NETs [7].
Management of PNETs associated with TSC remains controversial. The National Comprehensive Cancer Network recommends surgical resection if symptomatic or >2 cm, and selective resection for tumors 1–2 cm. It is unclear if TSC-associated PNETs behave similarly to sporadic ones [8].
A 2023 retrospective study compared perioperative outcomes and long-term results between TSC-associated and sporadic PNETs. TSC patients underwent surgery at younger ages and more frequently had multifocal tumors. None experienced recurrence during 6 years of follow-up [9].
Thus, the optimal surgical strategy for TSC-associated PNETs is still under investigation, and recent evidence supports more conservative approaches [9].
Conclusions
This case highlights the importance of maintaining high clinical suspicion for PNETs in patients with predisposing conditions.
Early diagnosis with active interventions and a multidisciplinary approach is essential for proper management and follow-up of these patients.
Ethical approval: Informed consent was obtained from the patient and family for publication of this case and related images.
Conflict of interest: The authors declare no conflicts of interest.
References
1. Dasari A, Shen C, Halperin D, Zhao B, Zhou S, Xu Y, et al. Trends in the Incidence, Prevalence, and Survival Outcomes in Patients With Neuroendocrine Tumors in the United States. JAMA Oncol. 2017 Oct 1;3(10):1335-1342. doi:10.1001/jamaoncol.2017.0589.
2. Bucheli AC, Balarezo PA, Velasquez JS. Pancreatic neuroendocrine tumors: a review article. Dom. Cien. 2023 Oct-Dec;9(4):1404-1420.
3. Zhang X, Zhong X, Lin X, Li X, Tian H, Chang B, et al. Tuberous Sclerosis Complex With Multiple Organ Tumors: Case Report and Literature Review. Front Oncol. 2022 Jul 19;12:916016. doi:10.3389/fonc.2022.916016.
4. Connor RF (Ed). Classification, clinical presentation, diagnosis, and staging of pancreatic neuroendocrine neoplasms. In: UpToDate. Wolters Kluwer. (Accessed May 2, 2025). Available at: https://www.uptodate.com/contents/classification-clinical-presentation-diagnosis-and-staging-of-pancreatic-neuroendocrine-neoplasms
5. Metz DC, Jensen RT. Gastrointestinal Neuroendocrine Tumors: Pancreatic Endocrine Tumors. Rev Gastroenterol Clin North Am. 2008 Nov;135(5):1469-1492. PMCID: PMC2612755. PMID: 18703061.
6. Pascual-Corrales E, Araujo-Castro M. Neuroendocrine tumors. Medicine. 2020;13(18):1019-28.
7. Somatostatin Analogs in Clinical Practice: A Review. Int. J. Mol. Sci. 2020;21:1682. doi:10.3390/ijms21051682.
8. Liu C, Lele SM, Goodenberger MH, Reiser GM, Christiansen AJ, Padussis JC. Malignant tumors in tuberous sclerosis complex: a case report and review of the literature. BMC Med Genomics. 2024 May 27;17(1):144. doi:10.1186/s12920-024-01913-8.
9. Shahzard A, Ventin M, Nebbia M. Long?Term Outcomes of Tuberous Sclerosis Complex?AssociatedNon?functional Pancreatic Neuroendocrine Tumors: Should We Be More Conservative? Ann Surg Oncol. 2023;30:7748–7755. doi:10.1245/s10434-023-14157-0.
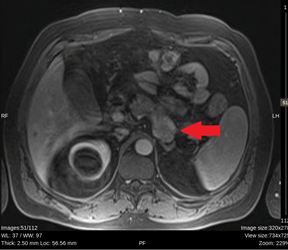
Figure 1