Bigeminal Rhythm in Acute Intracranial Haemorrhage
Bigeminal Rhythm in Acute Intracranial Haemorrhage
Dr. Ramachandran Muthiah *
*Correspondence to: Dr. Ramachandran Muthiah, President of All Nations, Morning star hospital, Enayam Thoppu, Kanyakumari District, India.
Copyright
© 2025 Dr Ramachandran Muthiah. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Received: 23 October 2025
Published: 01 November 2025
Abstract
Both ventricular bigeminy and ventricular fibrillation may occur in patients who have sustained intracranial hemorrhage. To salvage the patient, it is imperative that immediate and corrective treatment to be instituted. Cardiac arrhythmias occurring with intracranial lesions may be successfully managed by appropriate pharmacologic intervention and administration of atropine eliminated the rhythm abnormality due to an increase in intracranial pressure.
Key words
Intracranial hemorrhage, prominent U waves, intracranial pressure, ventricular bigeminy, atropine.
Bigeminal Rhythm in Acute Intracranial Haemorrhage
Introduction
Cardiac effects of intracranial hemorrhage was initially described in 1903 by Cushing, who noted alterations in blood pressure and cardiac rhythm in such patients [1]. Byer et al described ECG changes in a patient with subarachnoid hemorrhage [2]. Later a series of experiments described the effect of hypothalamic stimulation on ECG morphology and rhythm indicating that myocardial damage need not be present for ECG changes to occur [3],[4]. The distinctive abnormalities are prolonged QT intervals, large upright or deeply inverted T waves, bradycardia, prominent U waves and abnormalities of the ST segments [5]. The mechanism by which this occurs appears to be related to abnormal autonomic nervous system function, effects of catecholamines on the myocardial cells and can be prevented by vagolytics and adrenergic blockers. Echocardiography revealed no regional wall motion abnormalities and no LV dysfunction even though it was described in 10% of cases of intracranial hemorrhage [6], especially more frequent with women due to the difference in autonomic nervous system.
Case Report
50 years old female, a housewife was hospitalized with coma following head injury due to a fall in the bathroom accidentally. Both pupils constricted and reacting to light. Temperature was 37 ?C (98.6° F). The blood pressure was 260/130mmHg and refractory to appropriate antihypertensive drugs.
No abnormal blood pressure differences in limbs. Pulse rate was 58 bpm at the time of admission and suddenly reduced to 40 bpm. Respiration was regular and suddenly became deep and irregular. Neurologically, there was an exaggerated deep tendon reflexes with bilateral extensor plantar and early decerebrate rigidity. Hematological, liver and renal parameters were normal. Echocardiography revealed normal study. She was not taking any medications and not a known hypertensive or diabetic. She is an asian black, height 159 cm and weight is 65 kg. No other family members have similar episodes.The ECG at the time of admission shown in Figure (1) revealed prominent U wave due to slow heart rate. Subsequently, the patient developed further slowing of heart rate with deep, irregular respiration and ECG at this time shown in Figure (2) revealed ventricular bigeminy. Since this rhythm was slow rate induced, it was treated with atropine to sinus rhythm as shown in Figure (3). CT brain revealed intracranial hemorrhage with midline shift and hydrocephalus which may be the cause for the slowing of heart rate with ventricular bigeminy and refractory extreme hypertension.
Etiopathogenesis
Review of literature
The electrocardiographic abnormalities in localized cerebrovascular hemorrhage have been described. Frontal lobe hemorrhages are associated with QT prolongation and neurogenic T waves [7] (deep (>5 mm) or giant (> 10 mm) [8], widespread, symmetrically inverted T waves also known as ‘cerebral’ or ‘sympathetic’ T waves’) which may be a clinical vigilance for impending cerebrovascular events as shown in Figures 4 to 6 [9] & 7 and brain stem hemorrhage with noncardiogenic pulmonary edema.
The sudden development of atrial fibrillation may occur as a consequence of insular infarctions [11] since the autonomic regulation of cardiac rhythm constitutes an integrated relay system and its highest level of control is exerted by the cerebral cortex, particularly the insula. The role of inflammation in the genesis of poststroke AF within the first few days after ischemic stroke may occur through inflammatory mediators stimulating the intrinsic autonomic system and also by direct damage to atrial myocardium [12]. It is proposed that the cause of ECG abnormalities in association with lesions in the vicinity of Brodmann area 13 on the orbital surface of the frontal lobe, or around the circle of Willis, results from autonomic disturbances due to alterations in sympathetic and parasympathetic tone mediated by fibers from the orbito-frontal area to the heart via the stellate ganglia [13]. Widespread T-wave inversions with slight ST depression may occur secondary to subarachnoid haemorrhage [14].
Discussion
Elevations in blood pressure and its therapeutics
The human brain accounts for about 2% of body weight, receives 15%-20% of cardiac output and is responsible for approximately 20% of total body oxygen consumption. Much of this energy requirement is provided from glucose metabolism, which during its oxidative or aerobic process, promotes the generation of 38 molecules of adenosine triphosphate (ATP), in order to ensure the proper functioning of the Na+/K+ pump and promote ionic homeostasis [15]. “Spastic constriction of the arteries”, what today is called “essential” or “primary” hypertension, characterized by hypertrophy of the heart wall, arteries and arterioles before the blood pressure was measured in humans and it is called as ‘Bright disease’ in 1836 [16].
Most experts would accept that a BP of >200/120 mmHg is severe, and needs urgent attention, but the degree of urgency depends on the precise circumstances. For example, a BP of 180/100 mmHg in a poorly adherent, uncontrolled chronically hypertensive patient, would not usually be considered as acute severe HTN needing immediate treatment, whereas a BP of 160/100 mmHg would be a medical (hypertensive) emergency in the context of acute end organ damage (EOD), such as preeclampsia [17].
A transient elevation in arterial blood pressure is common in up to 84% of patients presenting with acute stroke [18], often associated with poor prognosis [19], and result from previous arterial hypertension but also from the physiological responses to brain injury [20]. Constant cerebral blood flow occurs over a mean blood pressure (MAP) of 50 to 150 mmHg [21]. [22] to 60 to 150 mmHg [23],[24],[25] in normotensives. The relationship of CBF (cerebral blood flow) with systemic arterial pressure remains essentially nonlinear, ie, CBF depends on ABP (arterial blood pressure), but does not increase or decrease in proportion to the changes of ABP. In patients with chronic hypertension without neurological deficits, the absolute level of cerebral blood flow is same as in healthy normotensive people as 50ml/100gm/minute and hypertension shifts it to a higher pressure [26] and loss of autoregulation ( inherent ability of blood vessels to keep the cerebral blood flow relatively constant over a wide range of activities and blood pressure levels) occurs at MAP of >150-160 mmHg in chronic hypertension [27]. In patients having acute stroke with extreme hypertension (very high (>220/110 mm Hg), it is justified to lower blood pressure cautiously as initially lowering by 15% [28].
Overtreatment below the autoregulatory limit in acute cerebrovascular event may reduce cerebral perfusion and aggravate brain damage. In patients with an intracerebral haemorrhage, intervention to control the blood pressure is required only when the systolic BP exceeds 170 mmHg and intensive lowering of systolic blood pressure to <140 mm Hg is proven safe [29] and < 130 mmHg in patients with chronic coronary and cerebrovascular diseases [30] for improving the functional outcome. During acute phase, patients may have resistant HTN (hypertension) due to sympathetic surge [31]. A few weeks later, they may require fewer medications and be at risk of hypotension unless the doses of medications are adjusted promptly.
Aspirin 300mg orally or rectally should be given as soon as possible if a haemorrhagic stroke has been excluded. Thrombolysis should only be given if administered within 4.5 hours of onset of stroke symptoms and haemorrhage has been definitively excluded (i.e. Imaging has been performed). Alteplase (t-PA) is currently recommended by NICE (National Institute for Health and Care Excellence) and blood pressure should be reduced to below 185/110 mmHg before alteplase [32]. Patients treated with thrombolysis should be started on an antiplatelet agent after 24 hours once significant haemorrhage has been excluded. Blood pressure (BP) should not be lowered aggressively in the acute phase of an ischemic stroke unless it is severely elevated (e.g., >220/120 mmHg) or there are complications like hypertensive encephalopathy, aortic dissection, acute kidney injury, acute coronary event, acute heart failure or preeclampsia/eclampsia) and these conditions may require an emergency BP reduction [33]. In acute aortic dissection, the systolic blood pressure should be reduced to 120 mm Hg within the first hour, with a target heart rate of around 60 bpm to decrease aortic shear stress and the first-line treatment is typically a beta blocker. In Acute myocardial infarction, nitroglycerin is the drug of choice for blood pressure management unless the patient has used phosphodiesterase inhibitors (eg, sildenafil or tadalafil) within the previous 48 hours. In pheochromocytoma crisis, a rapid reduction of systolic blood pressure to less than 140 mm Hg within the first hour is recommended and the preferred agents include phentolamine, clevidipine (Initial infusion of 1-2mg/hr), and nicardipine (5-15 mg/h). In preeclampsia and eclampsia, immediate blood pressure control is necessary and the first-line drugs include hydralazine, labetalol (0.25-0.5 mg/kg IV bolus; 2-4 mg/min), and nicardipine, typically in conjunction with magnesium sulphate for seizure prophylaxis [34].
Malignant hypertension is a severe and aggressive form of hypertensive emergency, defined by an extreme elevation of blood pressure with hypertension-associated microangiopathies affecting various organs, including the retina, kidneys, and brain [35]. The hallmark feature of malignant hypertension is retinal involvement, presenting with changes such as flame-shaped hemorrhages and papilledema. Patients may also present with hypertensive encephalopathy, pulmonary edema, and acute renal dysfunction. As the name indicates, malignant hypertension is associated with rapid disease progression and a poor prognosis, if not promptly and properly treated. Patients experiencing hypertension emergencies require admission, and oral antihypertensive medications are not recommended for initial management [36].
Hypertensive urgency is a form of hypertensive crisis characterized by a severe elevation in blood pressure, typically a systolic blood pressure greater than 180 mm Hg or a diastolic blood pressure exceeding 120 mm Hg, without evidence of acute or imminent target organ damage and do not require hospital admission [37]. These cases are often managed by adjusting existing antihypertensive therapy or adding oral agents and if not adequately treated, they remain at risk of developing a hypertensive emergency.
Diastolic pressure exceeding 130 mmHg is often associated with acute vascular damage and must be treated more rapidly with parenteral drugs. The prior preference for liquid nifedipine by mouth or sublingually has been deflated because of occasional ischemic complications from too rapid reduction in blood pressure. Intravenous furosemide is not indicated since volume depletion may aggravate vasospasm and increases cerebral ischemia. Predominant arterial vasodilators such as hydralazine and sodium nitroprusside are not usually preferred in the setting of intracranial hemorrhage.
If diastolic pressure exceeds 140mmHg and the patient has aortic dissection, a constant infusion of nitroprusside (0.25μg/kg/min) [38] is most effective. However, nitroprusside acts as a venous and arteriolar dilator so that venous return and cardiac output are lowered and intracranial pressure may increase. So other agents such as labetalol and esmolol are widely used in patients having hypertension with acute stoke. Intravenous calcium channel blockers (eg, nicardipine) and β-blockers (eg, labetalol) are the treatment of choice for early BP reduction.
For patients with large ICH (intracerebral hemorrhage) (volume > 30 cubic centimeters (cm3) or symptomatic perihaematoma oedema, it may be beneficial to keep serum sodium level at 140–150 mEq/L for 7–10 days to minimise oedema expansion and mass effect. Mannitol (0.5–1 g/kg) and hypertonic saline (HTS) can be used emergently for worsening cerebral oedema, elevated intracranial pressure (ICP) or pending herniation. HTS should be administered via central line as continuous infusion (3%) or bolus (23.4%). Ventriculostomy is indicated for patients with severe intraventricular haemorrhage, hydrocephalus or elevated ICP. Patients with large cerebellar or temporal ICH may benefit from emergent haematoma evacuation [39].
Interventional and surgical therapy
Intra-arterial clot retrieval (Mechanical thrombectomy) is safe and effective and mechanical thrombolytic devices can remove a clot in a matter of minutes, whereas pharmaceutical thrombolytics, even those delivered intra-arterially, may take as long as 2 hours to dissolve a thrombus and patients should undergo the procedure within 6 hours of symptom onset [40].
Decompressive hemicraniectomy is considered in patients with signs on CT of an infarct of at least 50% of the middle cerebral artery territory [41]. Since the extreme hypertension in this patient is refractory to appropriate antihypertensive agents and CT brain revealed midline shift with acute hydrocephalus, emergent ventriculostomy in the brain is to be done.
Neurogenic pulmonary edema
It is a clinical condition characterized by the acute accumulation of extravascular fluid in the lungs following a severe central nervous system (CNS) injury, most commonly involving the brainstem [42] and manifests as dyspnea, tachypnea, cough, rales, pink frothy sputum, hypoxemia and typically results as acute respiratory distress. W. T. Shanahan was the first to describe acute NPE (neurogenic pulmonary edema) in 1908 and Francois Moutier notably described the sudden onset of pulmonary edema among soldiers who sustained headshot wounds during World War I. Neurogenic pulmonary edema may have both a cardiogenic component, related to systemic hypertension, and a noncardiogenic (pulmonary capillary leak) component. One of the main proposed mechanisms is sympathetic storm/catecholamine-mediated pulmonary vasoconstriction. Catecholamine surge can cause neurogenic myocardial stunning, leading to transient left ventricular dysfunction, which exacerbates pulmonary congestion and edema called as Neurogenic myocardial injury (Takotsubo-like Cardiomyopathy) [43], [44]. Sympathetic overactivity shifts blood from the systemic to the pulmonary circulation, with a secondary elevation of left atrial and pulmonary capillary pressure. Thus, an imbalance of Starling forces (a hydrostatic mechanism) may be the basis for this form of pulmonary edema. Pulmonary capillary leak due to pressure - induced mechanical injury and/or direct nervous system control over capillary permeability may also play a role. Treatment consists of ventilatory support and manoeuvres to reduce intracranial pressure.
A chest x-ray (CXR) is essential for differentiating this condition from aspiration pneumonitis. With aspiration pneumonitis, the radiographic features (centrilobular nodules, often in a tree-in-bud pattern: reflect distal airway impaction of the aspirated particles [45]) will evolve over a few hours and can take up to 3 weeks to resolve in contrast with the alveolar infiltrates (nonspecific, bilateral, rather homogeneous airspace consolidative appearances with an apical predominance in 50% of cases [46]) seen in NPE, which typically occur immediately after the injury. Brain natriuretic peptide (BNP) and troponin levels may be measured to assess for concurrent cardiogenic causes [47].
Neurological deficits
Clinically, a more depressed level of consiousness without any fluctuating mental status or seizures and extreme refractory hypertension favour intracranial hemorrhage rather than metabolic or hypertensive encephalopathies and the key differentiating features are shown in Table 1
The most common location of a spontaneous intracerebral hemorrhage (ICH) is the putamen, which is usually associated with uncontrolled hypertension [48]. If intracranial pressure (ICP) increases, a Cushing response (hypertension, bradycardia, and bradypnea) may occur as compensation.
In subarachnoid hemorrhage, patients typically present with a thunderclap headache (sudden, severe headache that peaks within seconds to minutes), often described as the worst headache of their life and it is associated with meningeal irritation signs such as photophobia (increased sensitivity to light), neck stiffness [49] (meningismus- Inflammation of the meninges), nausea and vomiting (can be the signs of intracranial hypertension) and focal neurological deficits such as cranial nerve palsies (an oculomotor nerve palsy with pupillary dysfunction (pupil dilation from compression of the parasympathetic fibres that run on the outside of the third nerve), ptosis, and a ‘down and out’ eye) is associated with enlargement or rupture of the ipsilateral posterior communicating artery aneurysm and cerebral aneurysms may be associated with polycystic kidney disease), weakness, or seizures, often present either at the same time as a headache or soon thereafter, altered mental status ranging from mild confusion to coma, vision Changes diplopia (double vision) or pain around the eye, dizziness with mood and personality changes that include irritability. Subarachnoid hemorrhage patients may have a concurrent intraocular hemorrhage, known as Terson syndrome [50] and sometimes with symptoms of acute knifelike chest pain consistent with dissection of the vertebral artery, an unusual presentation and cause of spinal subarachnoid hemorrhage [51].
Noncontrast CT imaging of the brain is the standard imaging modality to detect the intracranial hemorrhage [52] during acute evaluation and contrast enhanced CT scan add specificity to detect subacute infarcts and to visualize venous structures. MRI is less sensitive than CT for detecting acute blood and it is used when the cause of intracranial hemorrhage is uncertain. Images of flowing blood in MRI (Magnetic Resonance Imaging) scan may identify AVMs (Arteriovenous malformations) as the cause of hemorrhage.
Electrolyte disturbances
Hyponatremia (a serum sodium level of less than 135 mEq/L (A "normal" serum sodium level, generally 136-145 mEq/L (or mmol/L)), categorized as mild (130–135 mEq/L), moderate (125–130 mEq/L), or profound (< 125mEq/L)) due to cerebral salt-wasting syndrome usually occurs in aneurysmal subarachnoid hemorrhage within first two weeks and significantly associated with increased risk of cerebral vasospasm and poor neurological outcomes [53]. The prevalence of hyponatremia in subarachnoid hemorrhage (SAH) was 30-55% [54] and occurs due to the inappropriate secretion of antidiuretic hormone (SIADH) [55].
Cerebral salt wasting (CSWS) is a disorder of sodium and water transport in the kidneys that occurs in patients with cerebral diseases due to increased activity of the sympathetic nervous system and dopamine release, leading to elevated renal pressure–natriuresis response and urinary sodium loss and release of brain natriuretic peptides by the injured brain. In symptomatic patients with acute hyponatremia (duration of < 48 hours), urgent correction by 4 to 6 mmol/L has to be initiated to prevent brain herniation and neurologic damage from cerebral ischemia [56]. For severe symptoms, intravenous infusion of 100 to 150 mL (2 mL/kg) of 3% NaCl over 15 to 20 minutes could be administered, repeated if necessary, to attain an increase in serum sodium values up to 2 mmol/L in the first hour and up to 4 to 5 mmol/L in 4 hours rapidly to control the seizures. In patients with mild to moderate symptoms, 3% NaCl is infused at 0.5 to 1 mL/kg/h.
Both CSWS and SIADH (syndrome of inappropriate antidiuretic hormone secretion) present with low serum osmolality, high urine osmolality, and a high urine sodium level. Basic difference between these two conditions is the amount of the extracellular fluid (ECF) volume. In CSWS, ECF loss will be seen along with urinary sodium loss, and hence patients will be in hypovolemic dehydrated state. Patients with SIADH may be either in euvolemic or hypervolemic condition. Management of CSWS patients [57] is directed at correcting the sodium loss and ECF loss and intravenous fluids, either isotonic or hypertonic saline, are used to correct intravascular volume and serum sodium depletion. Fludrocortisone, a potent mineralocorticoid (in doses of 0.1–1 mg/d), which causes reabsorption of sodium and water in the distal renal tubule, leading to expansion of the ECF, may be given in CSWS patients. Close monitoring of serum sodium levels is important to assess the extent of correction and prevent overcorrection leading to osmotic demyelination syndrome.
The normal serum osmolality is 275–295 mOsm/L. When it is normal or increased, the differentials include hyperlipidemia, hyperproteinemia, hyperglycemia, or mannitol infusions. The serum osmolality in CSWS or SIADH will be less. The normal random urine osmolality should average between 300 and 900 mOsm/L. Some cases of hypotonic hyponatremia may be due to primary polydipsia, and in such patients, urine osmolality will be less than 100 mOsm/L due to preserved kidney dilution capacity. The urine osmolality of CSWS and SIADH is usually high [58]. Urinary sodium will be low (< 20 mEq/L) in hypovolemia cases, whereas it will be elevated (> 40 mEq/L) in SIADH as well as in CSWS.
ECG changes
ECG abnormalities occur in 60-90% of patients with intra-parenchymal or subarachnoid bleed and in about 5-20% of patients with acute ischaemic stroke [59]. The ECG shown in Figure (1) revealed prominent U waves in midprecordial leads. Usually the U wave is about 0.1mV (10% of height of T wave) and follows the T wave. A normal U wave is usually upright and its voltage is typically less than 25% of the preceding T wave. It is not always visible on an ECG due to its low amplitude, but it is best seen in leads V2 and V3. U wave was considered significant if it was visible in more than 2 leads [60].
It is more easily observed in cases of bradycardia (slow heart rate) and produced by the repolarization of both purkinje fibres and M cells (mid-myocardial cells) [61] and corresponds to early after depolarization [62] and the exact U-wave's origin remains unknown and is subject to debate as theories
suggesting late depolarisation, delayed or prolonged repolarisation, stretch and intrinsic differences in the terminal AP [63]. A physiological U wave is thought to be due to delayed repolarization of the Purkinje system and pathological “U” wave as seen with hypokalemia is the consequence of electrical interaction among ventricular myocardial layers at action potential phase 3 of which repolarization slows [64]. Abnormal U wave was considered as negative U wave with more than 0.1mv depth or positive U wave higher than 25% of T wave. Slow heart rate, hypokalemia, hypomagnesemia, certain drugs such as quinidine, phenothiazines, tricyclic antidepressants and hypothermia which prolong the action potential duration of purkinje fibres and M cells are associated with prominent U waves. Negative U waves have been noted in patients with significant left ventricular ischemia and left ventricular hypertrophy and are also inadequately explained [65]. Exercise induced U wave inversion correlates with severe stenosis (>75 to 90%) of left anterior descending coronary artery [66],[67].
When potassium levels are <2.7 mmol/L, changes in the ECG include dynamic changes in T-wave morphology (T-wave flattening and inversion), ST-segment depression, and the appearance of U waves [68] which is the hallmark of significant hypokalemia and may become taller than T wave since the decreased extracellular potassium will result in hyperpolarized cells with increased action potential duration. Hypothermia is usually associated with prominent J wave (an abnormal, dome-like or humped elevation that appears at the junction of the QRS complex and the ST segment), when the body temperature drops below 35?C [69]. This distinctive feature is also known as Osborn wave or hypothermic hump and the degree of hypothermia correlates linearly with the amplitude of the Osborn wave [70]. In this ECG, the heart rate is low and the serum electrolytes and body temperature were normal. The prominent U wave with slower heart rate is significant and its amplitude is smaller than the T wave.
The PVC (premature ventricular contractions) is electrocardiographically defined as a premature QRS complex with an abnormal morphology and duration greater than 120 milliseconds. American Heart Association (AHA) and Heart Rhythm Society prefer the term "complexes" as electric activity does not always result in cardiac muscle contraction [71]. In certain instances, a QRS complex of duration less than 120 milliseconds can occur, leading to activation of both ventricles "synchronously" because of electrical stimulus from one of the fascicles through a specific conduction system [72]. A large T-wave classically follows it with discordant deflection (opposite polarity) from the QRS complex and typically, no preceding P waves are present. Left and right ventricular outflow tracts are the most common sites of origin of PVCs and account for approximately two-thirds of all PVC. PVCs with the morphology of a typical left bundle branch block (LBBB) and an inferior axis originate from the right ventricular outflow tract, while PVCs with the morphology of a standard right bundle branch block (RBBB) and an inferior axis originate from the left ventricular outflow tract. PVCs can originate from numerous other locations, eg, the ventricular free wall, ventricular septum, aortic cusp, tricuspid or mitral annuli, pulmonary artery, fascicles, or papillary muscles. Frequent PVCs can cause progressive left ventricular (LV) dysfunction and dilation [73], which may return to normal after successful catheter ablation of the PVCs [74]. Patients with a previous myocardial infarction with frequent PVCs are prone to sudden cardiac death. An "arrhythmic burden" occurs with >500 PVCs in 24 hours on Holter monitoring and frequent PVCs (>1000/day) are at risk of developing dilated cardiomyopathy [75] and for those with heart disease, PVCs are an indicator of increased mortality risk [76]. Cardiomyopathy is more likely in patients whose PVCs have a very wide QRS complex, the beats arise from the epicardium, or in patients with PVC occurring higher than a quarter of all beats on 24-hour Holter monitor (PVC burden of over 25%) and to be reversible after ablation [77]. If PVCs occur during recovery phase of exercise, the long-term mortality risk is higher [78].
PVCs are also definable by the pattern in which they occur relative to intrinsic beats. Bigeminy indicates a PVC following a single, standard QRS complex, whereas trigeminy indicates a PVC following two normal QRS complexes [79]. In the typical form of bigeminy, a ventricular premature beat is substituted for every alternate sinus beat, and each ventricular premature beat is followed by a compensatory pause. The impulse generated from the PVCs can often be propagated in a retrograde fashion to the atria and can cause a classic compensatory pause following the PVC. This pause is due to the atrioventricular (AV) nodal blockade due to depolarization from the retrograde impulse and subsequent refractory period of the AV node. Usually, a PVC is followed by a full compensatory pause because the sinus node timing is not interrupted [80] and thereby not affecting the heart rate. The compensatory pause must not be relied upon as the sole indicator of ventricular origin of a wide premature beat. In contrast, premature atrial contractions (PACs) are usually associated with an incomplete pause because the PAC usually enters the sinoatrial node and resets its timing, which enables the following sinus impulse to appear earlier than expected.
Not all PVCs, however, get followed by a pause. If a PVC occurs early enough, especially with prolonged heart rates, it may appear sandwiched between the two normal beats and this phenomenon is called an "interpolated PVC." The sinus impulse following the PVC is conducted with a longer PR interval because of the retrograde concealed conduction by the premature ventricular impulse into the AV-node junction, slowing the subsequent conduction of the sinus impulse. When the ventricular premature beat is “interpolated” between sinus beats, there is no pause, resulting in an almost doubling of the heart rate and a tachyarrhythmia with bigeminal pattern is produced (interpolated PVCs in bigeminy). Despite the doubled heart rate, the patient did not report any symptoms during the episode of interpolated ventricular bigeminy. When ventricular premature beats manifest different QRS- complex morphologies in the same lead, they are termed multiform ventricular premature beats and ventricular bigeminy occurs with constant coupling intervals but continually varying morphology. When the interval between the PVC and the normal beat is stable, it is termed fixed coupling. Variable coupling is when these intervals change from beat to beat. PVCs are considered "frequent" if they occur > 30 times per hour or > 20% of total heart beats [81]. Ventricular bigeminy typically has fixed coupling intervals, and each bigeminal beat looks identical to each other. Similarly, sustained VT is usually monomorphic with fixed intervals and morphology. When the bigeminy with fixed intervals but marked variation in morphology or a VT of constant cycle length but with a change in morphology, a single source of arrhythmia that has variable routes through which the rest of myocardial activation occurs, creating each exit’s own characteristic QRS complex [82].
Reentry produces a constant relationship between normal and premature beats and there are identical coupling intervals between each premature beat [83] and the preceding normal beat. Automaticity produces a varying relationship between normal and abnormal beats, but a constant relationship between abnormal beats and no identical coupling intervals between premature beats and normal beats with identical intervals between consecutive premature beats [84].
In the ECG shown in Figure 2, premature beats manifest same morphology in the same lead and there is identical coupling interval between each premature beat and the preceding sinus beat with a pause and this rhythm abnormality is ventricular bigeminy [85] due to reentry.
Low levels of serum potassium and magnesium are associated with higher prevalence rates of ventricular arrhythmias [86]. Life threatening arrhythmias may occur in the first day after the neurological events [87] and careful monitoring of potassium levels especially in patients with subarachnoid hemorrhage is warranted since hypokalemia is observed in up to 50% of patients with subarachnoid hemorrhage and this increases the likelihood of QT interval prolongation [88] and a torsades de pointes type of ventricular tachycardia. Normal QT interval varies inversely with heart rate and thus the corrected QT interval (QTc) was popularly used [89]. Spontaneous subarachnoid hemorrhage (SAH) causes a prolonged corrected QT interval (QTc) in 25% to 90% of patients [90] and also one of the most common electrocardiographic (ECG) abnormalities in patients with aneurysmal subarachnoid hemorrhage [91].
Enalapril or hydrochlorothiazide do not reduce, but metoprolol and diltiazem reduce premature ventricular complexes in patients with hypertension [92]. Intravenous lidocaine is generally the initial treatment of choice to suppress premature ventricular complexes in hospitalized patients especially in the setting of acute myocardial infarction [93]. Beta-adrenergic blockers are effective to decrease myocardial damage and in controlling both supraventricular and ventricular tachyarrhythmias associated with intracranial hemorrhage and head trauma, but can increase the likelihood of bradyarrhythmias [94]. In supraventricular arrhythmias, adenosine is a good treatment option in a patient where the AV node is the origin of arrhythmia; however, it can be catastrophic in a patient with a circuit formed by an accessory pathway. If adenosine is given to a patient with Wolff-Parkinson-White syndrome, the AV node will be blocked by the medications. However, the accessory pathway will remain uninhibited, which can lead to ventricular fibrillation and possible cardiac arrest[95][96] and so clinicians should be cautious in its use with patients who have WPW, making imperative the need for accurate diagnostic measures prior to treatment.
Both fast and slow heart rate can provoke the development of premature ventricular complexes [97]. Slowing the heart rate in some patients with sinus tachycardia can eradicate premature ventricular complexes. Increasing the basic heart rate with atropine or isoproterenol or by pacing, premature complexes accompanying slow ventricular rates can be abolished [98].
Since the rhythm in the ECG shown in Figure 2 is ventricular bigeminy and it may precede the onset of polymorphic ventricular tachycardia in the setting of intracranial hemorrhage and so it must be treated. Since the heart rate is slow, the bigeminal rhythm is rate induced rather than other causes. After intravenous atropine, the rhythm abnormality was abolished as seen in ECG shown in Figure 3.
Conclusion
The electrocardiographic abnormalities due to intracranial diseases are of clinical significance since they often mimic ischemic heart disease [99]. drugs, or electrolyte disorders. A significant correlation was established between the clinical manifestations of brainstem compression and premature ventricular contractions [100]. The occurrence of bigeminal and other cardiac arrhythmias is a manifestation of 'neurogenic stress cardiomyopathy', resulting from acute dysfunction and an imbalance of the autonomic nervous system. The bleeding and subsequent increase in intracranial pressure (ICP) can irritate or damage cardiovascular regulatory centers in the brain, such as those in the hypothalamus and brainstem and leads to a massive surge in circulating catecholamines (stress hormones), which directly damages myocardial tissue and causes various ECG changes and arrhythmias. The excess catecholamines can cause focal myocardial damage, such as subendocardial hemorrhages and myofibrillar degeneration, creating conditions for abnormal electrical activity and arrhythmias like bigeminy. The autonomic imbalance can also lead to prolongation of the QT interval, which increases the temporal dispersion of ventricular myocardial refractory times and raises the risk of life-threatening arrhythmias, including bigeminy and ventricular tachycardia/fibrillation.
Reference
1. Byer, E., Ashman, R., Toth, L.,A.(1947) Electrocardiograms with large, upright and long qt-intervals, American Heart Journal, 33, 796–806.
2. Cushing, H.(1903) The blood pressure reaction of acute cerebral compression illustrated by cases of intracranial hemorrhage, American Journal of Medical Sciences, 125, 1017–1044.
3. Weinberg, S., J., Fuster, J., M. (1960) Electrocardiographic changes produced by localized hypothalamic stimulations, Annals of Internal Medicine, 53, 332–341.
4. Attar, H.,J., Gutierrez, M., T., Bellet, S., Ravens, J.,R.(1963) Effects of stimulation of hypothalamic and reticular activating systems on production of cardiac arrhythmia, Circulation Research, 12:, 14–21.
5. Abildskov,J.,A., Miller, K., Burgess,M.,J., Vincent, W.(1970) The electrocardiogram and the central nervous system, Progress in Cardiovascular Diseases, 13, 2, 210-216.
6. Winder, K., Millar, V., C, Siedler, G., Knott, M., Dörfler, A., Engel, A., Achenbach, S., Hilz, M.,J., Kallmünzer, B., Schwab, S., Seifert, F., Fröhlich, K.(2023) Acute right insular ischaemic lesions and poststroke left ventricular dysfunction, Stroke and Vascular Neurology, 8, 4, 301-306.
7. Kajananan, Sivagurunathan & Subramaniyam, Merjiny & Jegathesan, Nalayini & Thampipillai, Peranantharajah (2025). Cerebral T waves in ischaemic stroke: an uncommon yet significant ECG finding, Asian Journal of Internal Medicine. 4. 45-46.
8. Cai, A., Chen, Z., Kang, F.(2023) Giant T-wave Inversion in an Older Patient With Sudden Loss of Consciousness, JAMA Internal Medicine, 183, 5, 482–483.
9. Daniel, M., Lindberg, M.,D., Edward, C., Jauch, M.,D., M.,S. (2006) Neurogenic T Waves Preceding Acute Ischemic Stroke, Images in Cardiovascular Medicine, Circulation, 114, 9, e369-e370
10. Rajkarnikar, R., Olenginski, G., Jacobs, S. et al.(2025) Cerebral T waves Preceding Acute Ischemic Stroke In A Patient Presenting With Syncope, JACC., Apr, 85 (12_Supplement) 3069.
11. Prats?Sánchez, L., Guisado?Alonso, D., Painous, C., Fayos, F., Pascual?Goñi, E., Delgado?Mederos, R., … & Martí?Fàbregas, J. (2017). Insular damage, new?onset atrial fibrillation and outcome after acute intracerebral hemorrhage, European Journal of Neurology, 25,3, 491-496.
12. Luciano, A., Sposato, MD, MBA, FAHA, Patricia, M., Riccio, MD, and Vladimir Hachinski, CM, MD, FRCPC, DSc (2014) Poststroke atrial fibrillation: Cause or consequence?,Critical review of current views, Neurology, 82 ,13, 1180-1186.
13. Beverly, J., Yamour, M.,R., Sridharan, Rice, J.,F., Flowers, N.,C.(1980) Electrocardiographic changes in cerebrovascular hemorrhage, American Heart Journal, 99, 3, 294-300.
14. Cadogan, M., Buttner, R., (2025) Raised Intracranial Pressure. ECG Library, Jan,14.
15. Michel Ferreira Machado1, Henrique Cotchi Simbo Muela, Valeria Aparecida Costa-Hong, et al (2021) Cerebral autoregulation in hypertensive patients: A review, Interventional Cardiology,.S4, 87-92
16. Folkow,B.,Nordlander, M.,K.(1982) Interaction between functional and structural elements in primary hypertension. In: Amery A, Fagard R, Lijnen P, Staessen J, editors. Hypertensive Cardiovascular Disease: Pathophysiology and Treatment, Dev Cardiovasc Med. 16: p.231-248.
17. Kulkarni, S., Glover, M., Kapil, V. et al (2023) Management of hypertensive crisis: British and Irish Hypertension Society Position document, Journal of Human Hypertension, 37, 863–879.
18. McManus, M., Liebeskind, D.,S.(2016) Blood Pressure in Acute Ischemic Stroke, Journal of Clinical Neurology, 12, 2, 137-146.
19. Dimiter, I., Hadjiev, Petya P. Mineva,(2023) Elevated blood pressure management in acute ischemic stroke remains controversial: Could this issue be resolved?, Medical Hypotheses, 80, 1, 50-52.
20. Philip M. Bath , DSc, FMedSci; Lili Song , MD, PhD; Gisele S. et al (2022) Blood Pressure Management for Ischemic Stroke in the First 24 Hours, Stroke, 53, 1074–1084.
21. N.A. Lassen (1959) Cerebral blood flow and oxygen consumption in man, Physiological Reviews, 39, 2, 183-238.
22. Gomez, J.,R., Bhende, B.,U., Mathur, R., Gonzalez, L.,F., Shah, V.,A.(2025) Individualized autoregulation-guided arterial blood pressure management in neurocritical care, Neurotherapeutics,22, 1, e00526.
23. Gottesman, R.,F., Egle, M., Groechel, R.,C., Mughal, A.(2024) Blood pressure and the brain: the conundrum of hypertension and dementia, Cardiovascular Research, 120, 18, 2360–2372.
24. Eames, P., Eames, P.,J., Blake, M.,J., Panerai,R.,B., Potter, J.,F.(2003) Cerebral autoregulation indices are unimpaired by hypertension in middle aged and older people , American Journal of Hypertension, 16, 9, 746–753,
25. Rickards, C.,A,, Tzeng, Y.,C.(2014) Arterial pressure and cerebral blood flow variability: friend or foe? A review, Frontiers in Physiology. Apr 7, 5, 120
26. Paulson, Olaf & Strandgaard, S & Edvinsson, L. (1990). Cerebral autoregulation, Cerebrovascular and brain metabolism reviews. 2. 161-192.
27. Fakhari, N., Aguet, J., Howell, A. et al (2025) Towards quantitative assessment of cerebrovascular autoregulation in human neonates using ultrafast ultrasound imaging. Scientific Reports, 15, 12374.
28. Steiner, T., Al-Shahi Salman, R., Beer, R., Christensen, H., Cordonnier, C., Csiba, L., et al (2014) European stroke organisation (ESO) guidelines for the management of spontaneous intracerebral hemorrhage, International Journal of stroke, 9, 840–855.
29. Mutimer, C.,A., Yassi, N., Wu, T.,Y. (2024) Blood Pressure Management in Intracerebral Haemorrhage: when, how much, and for how long? Current Neurology and Neuroscience Reports, 24, 7, 181-189.
30. Manolis, A.,J., Kallistratos, M.,S., Camafort, M., Coca, A. (2023) How low should blood pressure be in patients with chronic coronary and cerebrovascular diseases, European Journal of Internal Medicine, 109, 22-29.
31. Grassi, G., Quarti-Trevano, F., Cuspidi, C., Sanidas, E., Mancia, G., Thomopoulos, C. (2025) Sympathetic Overactivation in the Resistant Hypertensive Phenotype: A Meta-Analysis of Published Studies, Hypertension, 82, 8, 1346-1354.
32. Zonneveld, T.,P., Algra, A., Dippel, D.,W., Kappelle, L.,J., Van Oostenbrugge, R.,J., Roos, Y.,B., Wermer, M.,J., Van der Worp, H.,B., Nederkoorn, P.,J., Kruyt, N.,D.(2015) TRUTH investigators. The Thrombolysis in Uncontrolled Hypertension (TRUTH) protocol: an observational study on treatment strategy of elevated blood pressure in stroke patients eligible for IVT, BMC Neurology, Nov 23, 15, 241.
33. G?secki, D., Kwarciany, M., Kowalczyk, K., Narkiewicz, K., Karaszewski, B.(2020) Blood Pressure Management in Acute Ischemic Stroke, Current Hypertension Reports, 10, 23(1), 3.
34. Ahmed, I., Chauhan, S., Afzal, M.(2025) Hypertensive Crisis. [Updated 2025 Jun 22]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan-
35. Boulestreau, R., Van den Born, B.,H., Lip, G.,Y.,H., Gupta, A.(2022) Malignant Hypertension: Current Perspectives and Challenges, Journal of American Heart Association, Apr 05, 11(7), e023397.
36. Astarita, A., Covella, M., Vallelonga, F., Cesareo, M., Totaro, S., Ventre, L., Aprà, F., Veglio, F., Milan, A.(2020) Hypertensive emergencies and urgencies in emergency departments: a systematic review and meta-analysis, Journal of Hypertension, 38,7, 1203-1210
37. Khan, N.,N., Zurayyir, E.,J., Alghamdi, A.,M., Alghamdi, S.,F., Alqahtani, M.,A., Abdalla, E.,M., Jurays, N.,S., Alassiri, A.,M., Alzahrani, H.,A., Althabet, A.,A. (2024) Management Strategies for Hypertensive Crisis: A Systematic Review, Cureus,16, 8, e66694.
38. What is the initial pharmacological treatment for aortic dissection? (2025) Medical Advisory Board, Last updated: September 22.
39. Dastur, C.,K., Yu, W.(2017) Current management of spontaneous intracerebral haemorrhage, Stroke and Vascular Neurology, 24, 2(1), 21-29.
40. Jahan, R., Duckwiler, G.,R., Kidwell, C.,S., Sayre, J.,W., Gobin, Y.,P., Villablanca, J.,P., Saver, J., Starkman, S., Martin, N., Vinuela, F.(1999) Intraarterial thrombolysis for treatment of acute stroke: experience in 26 patients with long-term follow-up, AJNR American Journal of Neuroradiology, Aug;20(7),1291-1299.
41. Ali Mulhem (2024)Decision making for decompressive craniectomy (DC) in patients with malignant middle cerebral artery infarction (mMCAI) based on inclusion criteria of clinical studies: A systematic review, Brain Disorders, 15, 100161.
42. Al-Dhahir, M.,A., Hall, W.,A., Das, J.,M., et al.(2025) Neurogenic Pulmonary Edema. [Updated, Jul 7]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing.
43. Raja, H.,M., Herwadkar, A.,V., Paroutoglou, K., Lilleker, J.,B.(2018) Neurogenic pulmonary oedema complicating a lateral medullary infarct, BMJ Case Reports, Jul 26.
44. Kitagawa, T., Yamamoto, J., Kureshima, M., Maeda, H., Nishizawa, S.(2018) [Takotsubo Cardiomyopathy and Neurogenic Pulmonary Edema Following Fibrinolytic Therapy for Embolic Stroke:A Case Report], No Shinkei Geka, Neurological Surgery, Jan;46(1), 21-25.
45. Henry Knipe (2025) Acute aspiration pneumonitis, Radiopaedia, Aug 9.
46. Ashesh Ishwarlal Ranchod (2023) Neurogenic pulmonary edema, Radiopaedia, Apr 3.
47. Bonello, M., Pullicino, R., Larner, A.,J.(2017) Acute pulmonary oedema: not always cardiogenic, Journal of the Royal College of Physicians of Edinburgh, Mar;47(1), 57-59.
48. An, S.,J., Kim, T.,J., Yoon, B.,W.(2017) Epidemiology, Risk Factors, and Clinical Features of Intracerebral Hemorrhage: An Update, Journal of Stroke, Jan;19(1):3-10.
49. Tenny, S., Das; J.,M., Thorell, W. (2024) Intracranial Hemorrhage, [Updated 2024 Feb 17]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan.
50. Ziu, E., Khan Suheb, M.,Z., Mesfin, F.,B (2023) Subarachnoid Hemorrhage. [Updated 2023 Jun 1]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; 2025 Jan.
51. Barton, C.,W.(1988) Subarachnoid hemorrhage presenting as acute chest pain: a variant of le coup de poignard, Annals of Emergency Medicine, Sep;17(9):977-978.
52. Hillal, A., Ullberg, T., Ramgren, B. et al.(2022) Computed tomography in acute intracerebral hemorrhage: neuroimaging predictors of hematoma expansion and outcome, Insights Imaging, 13, 180.
53. Hulliel, A. F., Abuhashem, O. H., AlRabadi, A. M., Aldalki, S. K., Ahmad, O. A., Abdel Al, A. M., … Al Tah, B. (2025). Hyponatremia and cerebral vasospasm in subarachnoid hemorrhage: a systematic review and meta-analysis, Neurological Research, 1–12.
54. Chaovarin, C., Keandoungchun, P., Phinthusophon, A., Wangtanaphat, K., Veerasarn, S.(2024) Predictive factors of hyponatremia in subarachnoid hemorrhage and outcomes, Neurology Asia, 29(2), 321 – 328.
55. Kurokawa, Y., Uede, T., Ishiguro, M., Honda, O., Honmou, O., Kato, T., Wanibuchi, M.(1996) Pathogenesis of hyponatremia following subarachnoid hemorrhage due to ruptured cerebral aneurysm, Surgical Neurology, 46, 5, 500-508.
56. Verbalis, J.,G., Goldsmith, S.,R., Greenberg, A., et al (2013) Diagnosis, eval-uation, and treatment of hyponatremia: expert panel recommendations, American Journal of Medicine, 126(10, Suppl 1):S1–S4.
57. Momi, J., Tang, C.,M., Abcar, A.,C., Kujubu, D.,A., Sim, J.,J.(2010) Hyponatremia—what is cerebral salt wasting? Permanente Journal, 14(02), 62–65.
58. Rajendran, Adhithyan & Kannath, Santhosh & Vimala, Smitha & Rajan, Jayadevan. (2018) Cerebral Salt-Wasting Syndrome in Subarachnoid Hemorrhage: A Primer for Interventional Neuroradiologists, Journal of Clinical Interventional Radiology ISVIR. 02. 10.1055/s-0037-1609047.
59. Fentz, V., Gormsen, J.(1962) Electrocardiograph patterns in patients with acute cerebrovascular accidents, Circulation, 25, 22-28.
60. Nalabale, et al (2023) A Study of Analysis of Different Electrocardiographic Changes in Acute Cerebrovascular Accidents, International Journal of Pharmaceutical and Clinical Research, 15 (11), 1393-1399.
61. Watanabe, Y.(1975) Purkinje repolarization as a possible cause of the U wave in the electrocardiogram, Circulation, 51, 1030–1037.
62. El-Sherif, N.(1991) Early afterdepolarizations and arrhythmogenesis: experimental and clinical aspects, Arch Mal Coeur, 84, 227–234.
63. Kihlgren, M., Almqvist, C., Amankhani, F., Jonasson, L., Norman, C., Perez, M., Ebrahimi, A., Gottfridsson, C.(2023) The U-wave: A remaining enigma of the electrocardiogram, Journal of Electrocardiology, 79, 13-20.
64. Hlaing, T., DiMino, T., Kowey, P.,R., Yan, G.,X.(2005) ECG repolarization waves: their genesis and clinical implications, Annals of Noninvasive Electrocardiology, 10(2), 211-223.
65. Surawicz, B.(1998) U wave: facts, hypotheses, misconceptions, and misnomers, Journal of Cardiovascular Electrophysiology, 9, 1117–1128.
66. Costantini, M., Capone, S., Tondo, A., Oreto, G.(2008) Is exercise-induced U-wave inversion predictive of proximal left anterior descending coronary artery disease?, Journal of Electrocardiology, 41, 2, 99-101.
67. Raveendran, S., Hadfield, R., Petkar, S., Malik, N.(2012) Significance of exercise induced U wave inversion as a marker for coronary artery disease, BMJ Case Reports, Feb 10, bcr0420114132.
68. Wang, X., Han, D., Li, G (2020) Electrocardiographic manifestations in severe hypokalemia, Journal of International Medical Research, Jan;48(1):300060518811058.
69. Graham, C.,A., McNaughton, G.,W., Wyatt, J.,P (2001) The electrocardiogram in hypothermia, Wilderness & Environmental Medicine, 12, 232–235
70. Paurush Ambesh, MD, Gerald Hollander, MD and Jacob Shani, MD (2017) Osborn waves of hypothermia, Cleveland Clinic Journal of Medicine, 84 (10), 746-747.
71. Marcus, G.,M.(2020) Evaluation and Management of Premature Ventricular Complexes, Circulation, Apr 28, 141(17), 1404-1418.
72. Shi, L.,R (1983) Clinical significance of the site of origin of premature ventricular contractions (PVC)]. Zhonghua Xin Xue Guan Bing Za Zhi. 1983 Mar;11(1):36-38
73. Chugh, S.,S., Shen, W.,K., Luria, D.,M., Smith, H.,C.(2000) First evidence of premature ventricular complex-induced cardiomyopathy: a potentially reversible cause of heart failure, Journal of Cardiovascular Electrophysiology, Mar;11(3), 328-329.
74. Takemoto, M., Yoshimura, H., Ohba, Y., Matsumoto, Y., Yamamoto, U., Mohri, M., Yamamoto, H., Origuchi, H.(2005) Radiofrequency catheter ablation of premature ventricular complexes from right ventricular outflow tract improves left ventricular dilation and clinical status in patients without structural heart disease, Journal of American College of Cardiology, Apr 19, 45(8), 1259-165.
75. Panizo, J.,G., Barra, S., Mellor, G., Heck, P., Agarwal, S (2018) Premature Ventricular Complex-induced Cardiomyopathy, Arrhythmia & Electrophysiology Review, Jun;7(2), 128-134
76. Massing, M.,W., Simpson, R.,J., Rautaharju, P.,M., Schreiner, P.,J., Crow, R., Heiss, G.(2006) Usefulness of ventricular premature complexes to predict coronary heart disease events and mortality (from the Atherosclerosis Risk In Communities cohort), American Journal of Cardiology, Dec 15, 98(12), 1609-1612.
77. Sattar, Y., Hashmi, M.,F..(2025) Premature Ventricular Complex. [Updated 2025 Feb 16]. In: StatPearls [Internet]. Treasure Island (FL): StatPearls Publishing; Jan.
78. Lee, V., Perera, D., Lambiase, P.(2017) Prognostic significance of exercise-induced premature ventricular complexes: a systematic review and meta-analysis of observational studies, Heart Asia, 9(1), 14-24.
79. Koester, C., Ibrahim, A.,M., Cancel, M., Labedi, M.,R.(2020) The Ubiquitous Premature Ventricular Complex, Cureus, Jan 7, 12(1), e6585.
80. Lindow, T., Niklasson, A., Manna, D., Pahlm, O.(2018) Bigeminal sandwiches galore, Annals of Noninvasive Electrocardiology, Mar;23(2), e12484.
81. Yang, J., Dudum, R., Mandyam, M., Marcus, G (2014) Characteristics of unselected high-burden premature ventricular contraction patients, Pacing and Clinical Electrophysiology, 37, 1671-1680.
82. Samuel, J., Asirvatham, MD, and William, G., Stevenson, MD (2015)Reciprocating Reentry, Circulation: Arrhythmia and Electrophysiology: Teaching Rounds in Cardiac Electrophysiology, 8, 1512-1513.
83. De Vries, L.,J., Martirosyan, M., Van Domburg, R.,T., Wijchers, S.,A., Géczy, T., Szili-Torok, T.(2018) Coupling interval variability of premature ventricular contractions in patients with different underlying pathology: an insight into the arrhythmia mechanism, Journal of Interventional Cardiac Electrophysiology, Jan 51(1), 25-33.
84. Antzelevitch, C., Burashnikov, A.(2011) Overview of Basic Mechanisms of Cardiac Arrhythmia, Cardiac Electrophysiology Clinics, Mar 1, 3(1), 23-45.
85. Bigeminy (2023), Cleveland Clinic, 5 Jan
86. Tsuji, H., Venditti, F.,J., Jr, Evans, J.,C., Larson, M.,G., Levy, D.(1994) The associations of levels of serum potassium and magnesium with ventricular premature complexes (the Framingham Heart Study), American Journal of Cardiology, Aug 1, 74(3), 232-235.
87. Bernd Kallmünzer, MD, Lorenz Breuer, MD, Nicolas Kahl (2012) Serious Cardiac Arrhythmias After Stroke: Incidence, Time Course, and Predictors—A Systematic, Prospective Analysis, Stroke, 43, 2892-2897.
88. Fukui, Shinji & Katoh, Hiroshi & Tsuzuki, et al (2003). Multivariate analysis of risk factors for QT prolongation following subarachnoid hemorrhage, Critical care (London, England). 7. R7-R12. 10.1186/cc2160.
89. Johannesen, L., Garnett, C., Malik, M.(2014) Electrocardiographic data quality in thorough QT/QTc studies, Drug Safety, 37 3, 191–197.
90. Collier, Bryan & Miller, S & Kramer, Gary & Balon, Jennifer & Gonzalez, Luis. (2004) Traumatic Subarachnoid Hemorrhage and QTc Prolongation, Journal of neurosurgical anesthesiology, 16. 196-200.
91. Zhang, X., Lei, Y., Nan, L. et al.(2024) QTc prolongation after aneurysmal subarachnoid hemorrhage might be associated with worse neurologic outcome in patients receiving microsurgical clipping or embolization of the intracranial aneurysms: a retrospective observational study, BMC Neurology, 24, 170.
92. Papademetriou, V., Narayan, P., Kokkinos, P.(1994) Effects of diltiazem, metoprolol, enalapril and hydrochlorothiazide on frequency of ventricular premature complexes, American Journal of Cardiology, Feb 1, 73(4), 242-246.
93. Merino, J.,L., Tamargo,J., Blomström-Lundqvist, C., Boriani, G., et al (2025) Practical compendium of antiarrhythmic drugs: a clinical consensus statement of the European Heart Rhythm Association of the European Society of Cardiology, EP Europace, 27, I 8, euaf076.
94. Alali, A.,S., Mukherjee, K., McCredie, V.,A., et al (2017) Beta-blockers and Traumatic Brain Injury: A Systematic Review, Meta-analysis, and Eastern Association for the Surgery of Trauma Guideline, Annals of Surgery, 266(6), 952-961.
95. Brubaker, S., Long, B., Koyfman, A.(2018) Alternative Treatment Options for Atrioventricular-Nodal-Reentry Tachycardia: An Emergency Medicine Review, Journal of Emergency Medicine, 54(2), 198-206.
96. Rajkumar, C.,A., Qureshi, N., Ng, F.,S., Panoulas, V.,F., Lim, P.,B.(2017) Adenosine induced ventricular fibrillation in a structurally normal heart: a case report, Journal of Medical Case Reports, 22, 11(1), 21.
97. Engel, G., Cho, S., Ghayoumi, A., Yamazaki, T., Chun, S., Fearon, W.,F., Froelicher, V.,F..(2007) Prognostic significance of PVCs and resting heart rate, Annals of Noninvasive Electrocardiology, 12(2), 121-129.
98. Osakwe, A., Wightman, N., Deyell, M.,W., Laksman, Z., Shrier, A., Bub, G., Glass, L., Bury, T.,M. (2025) Dependence of premature ventricular complexes on heart rate—it’s not that simple, Journal of the American Medical Informatics Association, ocaf069.
99. McKamy Smith (1972)Ventricular bigeminy and quadrigeminy occurring in a case of subarachnoid hemorrhage, Journal of Electrocardiology, 5, 1, 78-85.
100. Stober, T., Sen, S., Anstätt, T., Bette, L.(1988) Correlation of cardiac arrhythmias with brainstem compression in patients with intracerebral hemorrhage, Stroke, 19(6), 688-692.
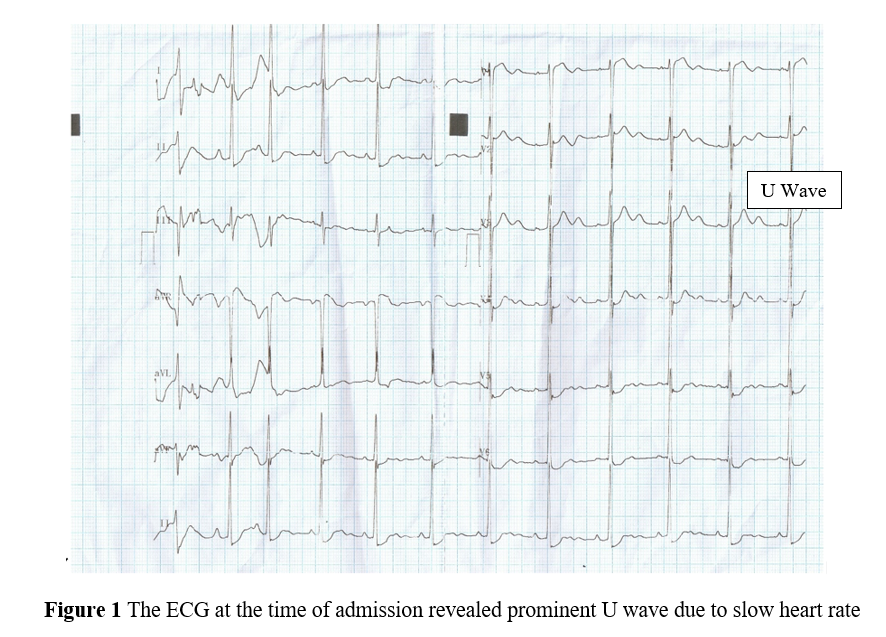
Figure 1
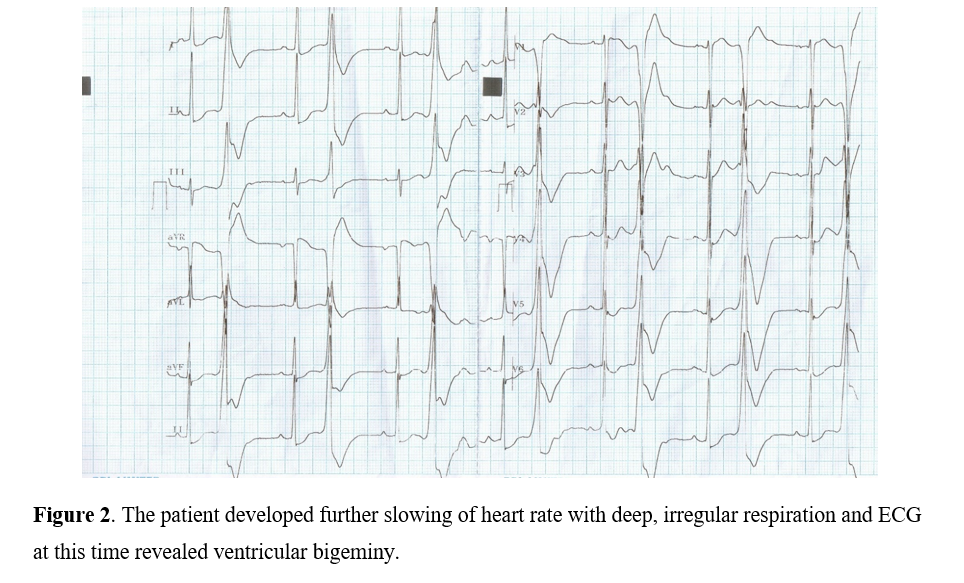
Figure 2
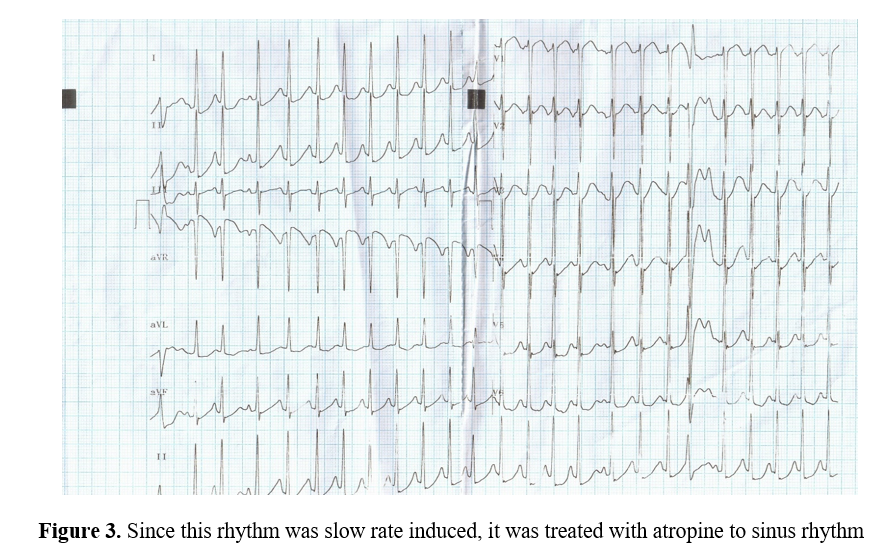
Figure 3
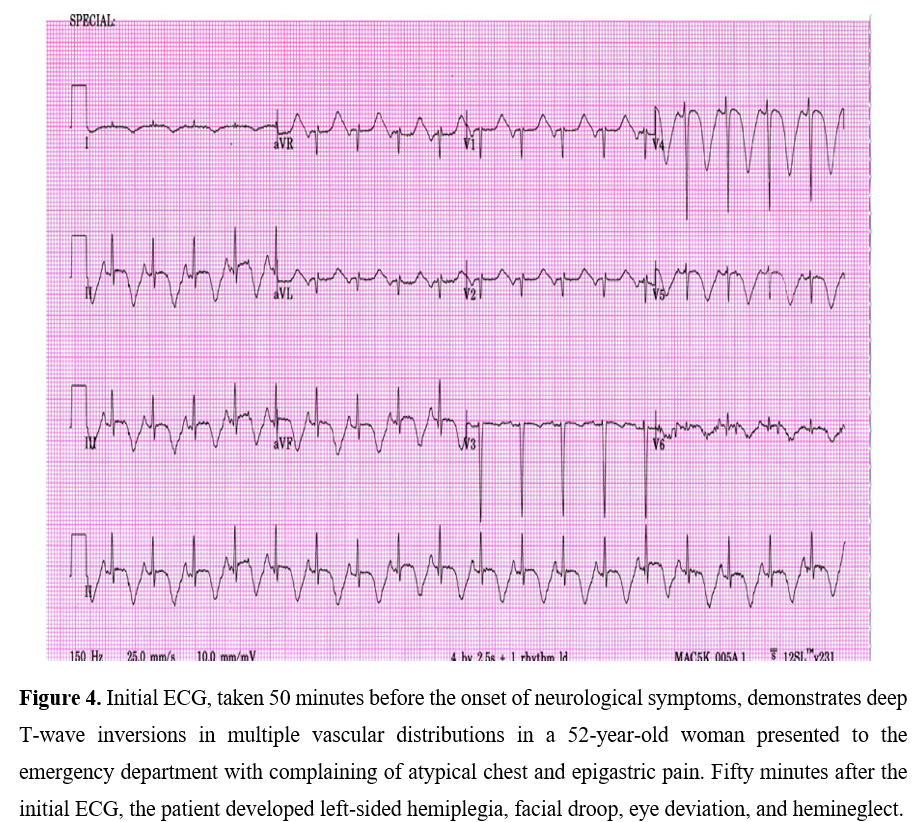
Figure 4
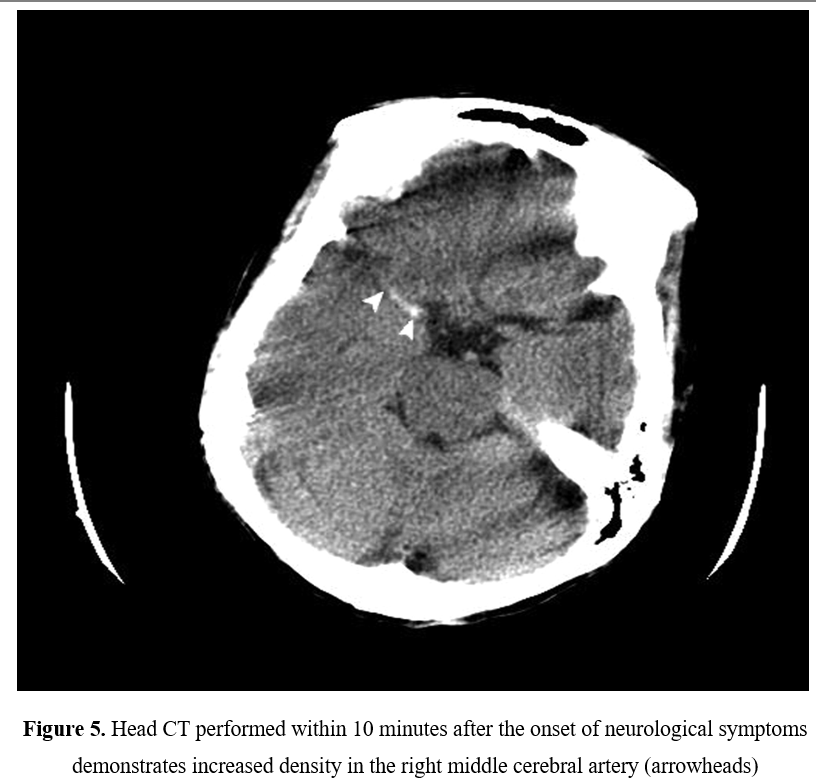
Figure 5
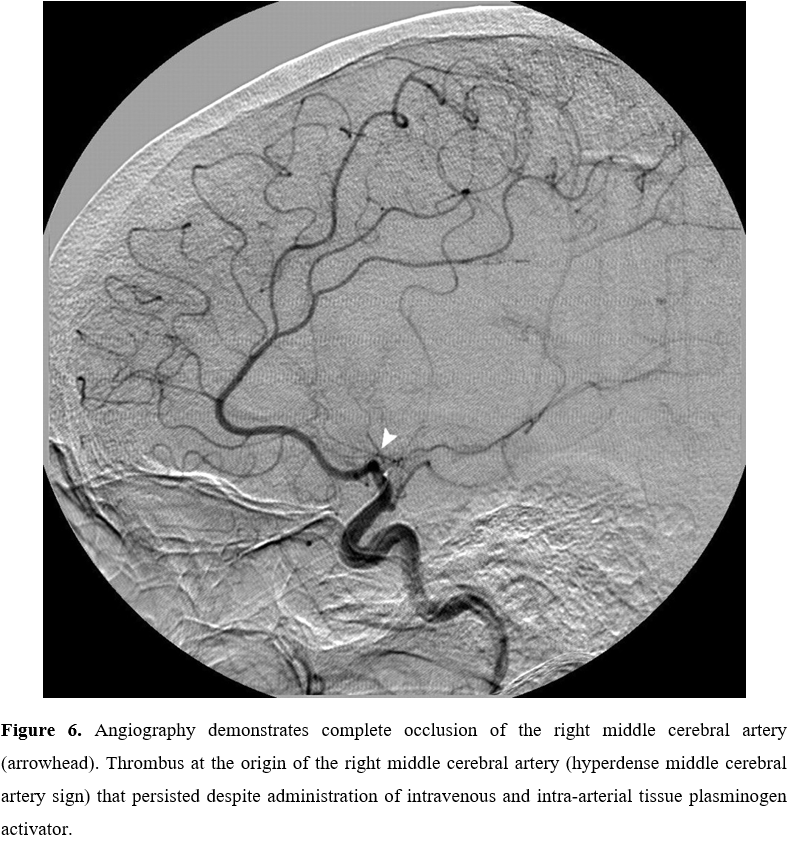
Figure 6
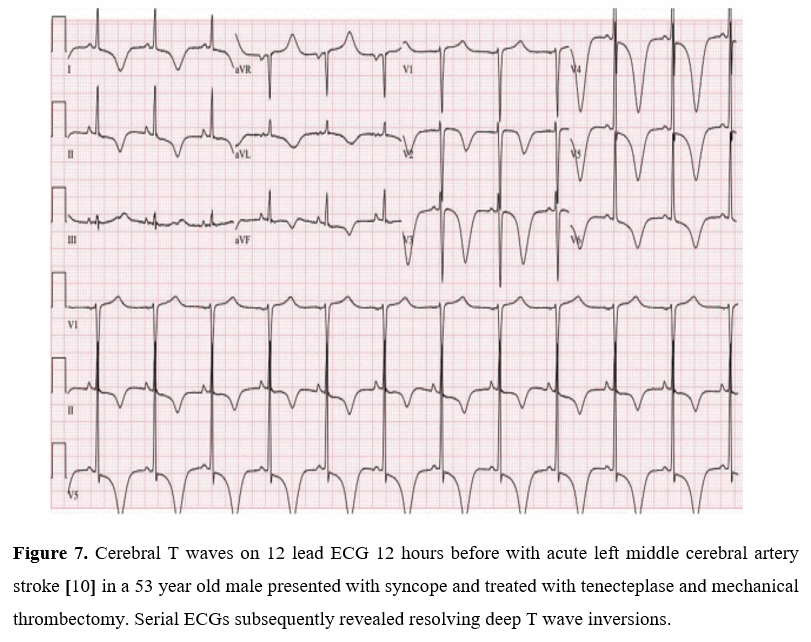
Figure 7
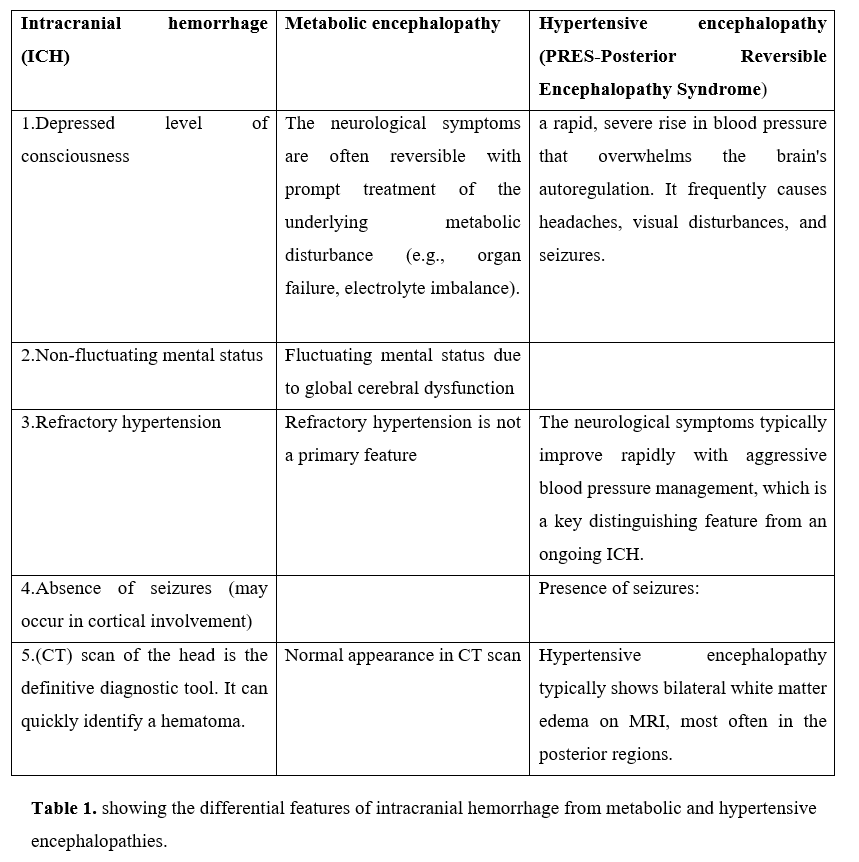
Figure 8